
|
|
资料
团标 | T/CAMDI 058-2020 最终灭菌医疗器械包装—GB/T 19633.1和GB/T 19633.2应用指南发表时间:2024-10-14 10:35 中国医疗器械行业协会
最终灭菌医疗器械包装—GB/T 19633.1和GB/T 19633.2应用指南 Packaging for terminally sterilized medical devices—Guidance on the application of GB/T 19633.1 and GB/T 19633.2
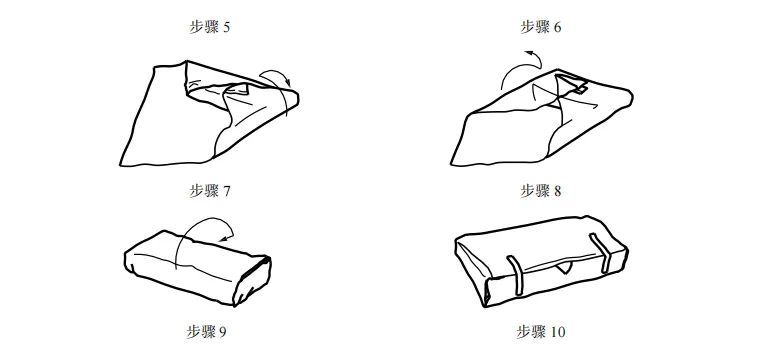
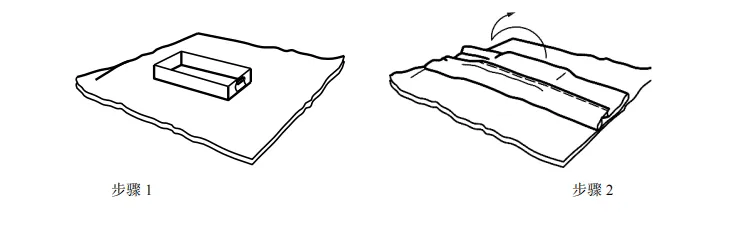
目 次 前言 引言 1 范围 2 规范性引用文件 3 术语和定义 4 医疗机构指南 5 工业指南 附录A(资料性)包装材料和无菌屏障系统的选择、评估和测试 — 工业和医疗机构指南 附录B(资料性)灭菌注意事项 — 工业和医疗机构指南 附录C(资料性)包裹方法实例 — 医疗机构指南 附录D(资料性)确认计划文件 — 医疗机构指南 附录E(资料性)安装鉴定文件 — 医疗机构指南 附录F(资料性)运行鉴定文件 — 医疗机构指南 附录G(资料性)性能鉴定文件 — 医疗机构指南 附录H(资料性)应对最坏情况的要求 — 工业和医疗机构指南 附录I(资料性)建立最终包装系统确认方案 — 工业指南 附录J(资料性)设计输入 -医疗器械特性 — 工业指南 附录K(资料性)风险分析工具 — 工业和医疗机构指南 附录L(资料性)抽样计划注意事项 — 医疗机构指南 附录M(资料性)稳定性试验(GB/T 19633.1-2015,6.4)— 工业指南 附录N(资料性)互联网使用 — 工业和医疗机构指南 附录O(资料性)试验方法确认 — 工业指南 附录P(资料性)合同包装商的使用 — 工业和医疗机构指南 附录Q(资料性)过程参数建立指南 — 工业指南 附录R(资料性)故障调查 — 工业和医疗机构指南 附录S(资料性)包装生产过程和包装系统设计可行性评估 — 工业指南 参考文献 前 言 请注意本文件的某些内容可能涉及专利。本文件的发布机构不承担识别这些专利的责任。 本文件由中国医疗器械行业协会提出。 本文件由中国医疗器械行业协会医疗器械包装专业委员会归口。 本文件起草单位:上海微创医疗器械(集团)有限公司、首都医科大学附属北京安贞医院、杜邦(中国)研发管理有限公司、南微医学科技股份有限公司、安姆科集团-毕玛时软包装(苏州)有限公司、上海建中医疗器械包装股份有限公司、东莞市安保医用包装科技有限公司。 本文件主要起草人:李然、周俊蕾、王瑾、秦蕾、钱军、丁艳琴、李宁、邓永忠、王清、汪友琼、宋翌勤、王燕、陈佩。 本文件首次发布于2020年12月。 前 言 无菌屏障系统需要在开封使用前确保内容物的无菌性,并确保无菌取用。 无菌屏障系统可根据处理、流通和贮存条件为灭菌医疗器械提供充分保护。对于需要重复处理的已包装及灭菌的器械,可能需要其他保护性包装与无菌屏障系统相结合形成包装系统。 在选择和实施前,组织应评估各无菌屏障系统或包装系统的性能,确保满足灭菌、贮存和处理条件的要求。管理无菌产品的组织应有贮存、处理和运输无菌产品的成文培训计划。 质量管理体系及其他要求存在地区性差异,这些地区性差异可能影响人力资源管理方式。但是,在任何情况下良好的培训过程都十分重要,各组织应确保其人员意识到包装和灭菌工作对患者安全的相关性和重要性。 GB/T 19633.1规定了材料、无菌屏障系统和包装系统的要求,包括包装系统设计和设计评估。GB/T 19633.2 规定了包装过程确认的要求。这两个文件为医疗器械保护、灭菌能力、保持无菌包装完整性以及无菌取用提供了要求。这些文件适用于医疗机构以及工业,且使用情况存在差异。 本文件为医疗机构(见第4章)和工业(见第5章)提供信息。可通过本文件更好地理解GB/T 19633.1和GB/T 19633.2的要求,并说明了满足这些文件要求的各种可行方式和方法。不需要使用本文件来证明符合GB/T 19633 的要求。 1 范围 本文件为应用 GB/T 19633.1 和 GB/T 19633.2 中的相关要求提供了指南,并未添加或以其他方式修改 GB/T 19633.1 和 GB/T 19633.2 的要求。 本文件提供评估、选择和使用包装材料、预成型无菌屏障系统、无菌屏障系统和包装系统的指南。为成型、密封和装配过程的确认要求提供指南。 本文件未提供打开后的包装材料及系统的应用指南。用于其它目的的包装(例如,在“无菌区”使用或运输被污染物),参见相关法规标准。 2 规范性引用文件 下列文件中的内容通过文中的规范性引用而构成本文件必不可少的条款。其中,注日期的引用文件,仅该日期对应的版本适用于本文件;不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。 GB/T 19633.1-2015 最终灭菌医疗器械包装 第1部分:材料、无菌屏障系统和包装系统的要求 GB/T 19633.2-2015 最终灭菌医疗器械包装 第2部分:成形、密封和装配过程的确认的要求 3 术语和定义 GB/T 19633.1-2015、GB/T 19633.2-2015和GB/T 19971-2015的术语和定义适用于本文件。 3.1 包装系统 packaging system 无菌屏障系统和保护性包装组合。 注:包装系统包括无菌屏障系统和保护性包装。但是,若无菌屏障系统可以保护医疗器械,便于无菌取用,并且具有足够韧性无需额外保护性包装时,无菌屏障系统还将满足包装系统的要求。不是始终都需要保护性包装,但必须保证在所有情况下都提供无菌开启/无菌取用。 3.2 保护性包装 protective packaging 为防止无菌屏障系统和其内装物从其装配直到最终使用的时间段内受到损坏的材料结构。 注 1:国家或地区法规可能要求使用保护性包装以避免污染手术环境。这些法规还可能要求在无菌屏障系统进入手术环境前拆除保护性包装。 注 2:保护性包装保护无菌屏障系统及其内容物。例如,防尘罩、包装盒、运输托盘。 3.3 无菌屏障系统 (SBS)sterile barrier system 防止微生物进入并能使产品在使用地点无菌取用的最小包装。 3.4 预成型无菌屏障系统 preformed sterile barrier system 已完成部分装配供装入和最终闭合或密封的无菌屏障系统。 示例:纸袋、组合袋和敞开着的可重复使用的容器。 注:预成型无菌屏障系统有各种类型。上述实例并未包括所有类型。 4 医疗机构指南 4.1 总则 请从包装材料和/或医疗器械制造商处获得书面指示说明,以了解有关无菌屏障系统的灭菌和后续 维持无菌状态方面之建议。 4.2 试验方法 GB/T 19633.1 和 GB/T 19633.2 所述试验方法指南参见本文件中医疗机构相关附录。 4.3 GB/T 19633.1 的符合性指南 a) 使用满足GB/T 19633.1要求的已知可追溯的材料(见GB/T 19633.1-2015, 5.1.3, 5.1.4和5.1.5所述要求); b) 无毒性,相关指南见A.3.3(见GB/T 19633.1-2015, 5.1.6所述要求); 注:若无菌屏障系统或相关部件含有天然乳胶,无菌屏障系统的标签中需注明含天然乳胶。 c) 在试验条件下进行灭菌、处理、流通、运输和贮存时,形成防止微生物进入的证明文件(见GB/T 19633.1-2015, 5.1.6和5.2所述要求); d) 宜证明满足材料和闭合处所需的物理性能要求(例如,重量或等级,密封宽度和密封强度),抗撕裂或抗穿刺能力,连续均匀开启或剥离特性,不会造成分层撕裂(见GB/T 19633.1-2015,5.1.7和5.1.9所述要求); e) 与无菌医疗器械的预期灭菌过程和参数相适应(见GB/T 19633.1-2015, 5.3所述要求); f) 与标识系统相适应;如有标识,在接触预期灭菌过程后,标识不褪色,印刷油墨不得降解、褪色或模糊不清(见GB/T 19633.1-2015, 5.4所述要求); g) 防止在贮存过程中受到环境条件的影响(例如,相对湿度、直接光照或荧光光照、温度)(见GB/T 19633.1-2015, 5.5和第7条所述要求); 注:材料或预成型无菌屏障系统制造商宜提供保存条件和有效期或保质期建议。若预期或实际保存条件超出建议要求,则咨询制造商。 h) 宜便于无菌取用。 注1:医疗器械和/或包装系统制造商需提供无菌取用说明。 注2:互联网是查找材料信息的有力工具,见附录N。 4.3.2 包装系统设计与开发指南(GB/T 19633.1-2015, 6.1 和 6.2) 4.3.2.1 选择准则 医疗机构确定包装系统时,宜考虑包装系统的设计和开发指南(见GB/T 19633.1-2015, 6.1和6.2所述要求)。医疗机构使用合同包装供应商或灭菌服务商时,需要考虑其它因素(见附录P)。 包装材料和包装系统选择: a) 制造商说明医疗包装应用的预期用途; b) 制造商提供技术信息,确认满足GB/T 19633.1中的材料相关要求; c) 在规定的预期贮存和运输条件下,在医疗器械使用前为医疗器械提供充分保护; d) 允许灭菌并与预期灭菌过程相适应,并且能够承受确定的灭菌条件; 注:并不是所有材料都适用于所有灭菌过程。与指定灭菌过程相适应的信息一般由医疗器械和/或包装系统制造商提供。关于一般灭菌过程的更多问题解释见附录B。 e) 在使用前保持无菌屏障的完整性; f) 确保使用时的无菌取用; g) 考虑使用有“打开迹象”的闭合方式; h) 便于识别内容物。 包装材料使用者确保无菌屏障系统或包装系统符合GB/T 19633.1的要求,满足有关产品相容性的要求,确保包装、灭菌、贮存和流通过程经过确认和得到控制。 4.3.2.2 考虑因素 医疗机构的选择包括评估无菌屏障系统和保护性包装(如适用)在使用前保持无菌屏障系统完整性的能力,以及在使用时允许无菌取用的能力。 包装组件的选择将取决于与医疗器械相关的风险,其使用条件、贮存和运输要求以及医疗程序,医疗机构应分析这些风险并制定降低/控制风险的程序(见附录 K)。 选择最适合的无菌屏障系统和/或包装系统材料时,考虑以下方面: a) 贮存条件和时长会影响所需无菌屏障系统或包装系统的类型选择。某些产品在使用前可能会贮存一段时间,可能需要更耐用的无菌屏障系统和/或额外保护性包装。无菌屏障系统或包装系统经受处理次数越多,发生破裂、盖子变形、垫圈受损、撕裂、穿孔或材料分离的概率就越大; b) 考虑待灭菌产品的尺寸、重量和形状。有些产品需要更耐用或更柔软的无菌屏障系统; c) 若使用多种包装组件,则必须验证组件之间、组件与组件内部的产品以及预期灭菌过程的相容性; d) 考虑运输方式和条件。在某些情况下会在医疗机构内部路线运输,也可能在不同医疗机构之间运输。包装系统暴露在不受控环境中,可能显著增加包装完整性破坏、危害无菌开启或污染内容物的风险。 4.3.2.3 装配注意事项 1) 由制造商声明无毒性,预期用于医疗包装; 2) 在贮存和运输过程中为医疗器械提供保护,直到器械最终使用; 3) 考虑与预期灭菌过程相适应,并能够承受确定过程的条件; 注:并不是所有材料都适用于所有灭菌过程。与指定灭菌过程的适宜性信息一般由制造商提供。关于常见灭菌过程挑战的进一步解释,见附录B。 e) 包装系统及其内容物的重量不宜超过国家有关人工搬运的规定。 4.3.2.4 标识注意事项 4.3.2.5 法规注意事项 在选择无菌屏障系统和/或包装系统的过程中宜考虑国家或相关地区的法规要求。 4.3.2.6 无菌屏障系统的通用选择 4.3.2.6.1 概述 无菌屏障系统的制造主要通过以下方式(但不限于)生产: ——可密封组合袋和卷材; ——灭菌包裹材料; ——可重复使用容器。 使用这些包装时宜考虑4.3.2.6.2~4.3.2.6.4的方面。 4.3.2.6.2 可密封组合袋和卷材(预成型无菌屏障系统) 可密封组合袋和卷材一般通过两种形式采购: ——沿两边密封的连续卷式或卷筒式。将卷展开并裁切成所需的长度,医疗器械置于上下两层之间,两端密封; ——组合袋是已裁切至规定尺寸,并完成三边密封的袋子。把医疗器械放入组合袋内,并对第四边密封。 考虑以下方面: 4.3.2.6.3 灭菌包裹材料 灭菌包裹材料有多种尺寸和重量,适宜各种应用。它可以是一次性或可重复使用织物形式。宜仔细考虑被包装产品及采用的包装技术。灭菌包裹材料可以用于包裹单独医疗器械或装入仪器盒、盒子或托盘中的医疗器械。 考虑以下方面: a) 根据待包装医疗器械的尺寸、形状和重量,或者医疗机构的指南和包装制造商的使用建议选择灭菌包裹材料的重量; b) 灭菌包裹材料的尺寸充分覆盖包装的医疗器械。必须牢固包裹包装的医疗器械,防止形成孔隙、翻滚和空气包。避免包装过紧导致的包装穿孔或撕裂。灭菌包裹材料还宜足够大,便于包裹材料适应灭菌循环挑战,防止撕裂。选择灭菌包裹材料时,包裹材料宜足以覆盖医疗器械,但不宜太大,以至于医疗器械包裹多次,从而影响灭菌介质渗透; c) 正确的包裹方法可以形成一个曲折路径,防止微生物进入无菌屏障系统。宜使用经制造商证明有效并推荐使用的包裹方法(见4.3.2.1)。选择的包裹方法宜便于医疗器械的无菌取用。医疗机构宜根据国家或地区法规在机构内对包裹方法进行验证或确认。可适用的包裹方法的国家标准或专业指南见附录C; d) 无菌包裹方法的设计宜使打开后的包裹材料避开无菌区域; e) 包裹材料操作台表面宜平整光滑,尺寸合适,良好光照且洁净; f) 包裹包装的设计宜确保各边缘严实,便于无菌取用并转移至无菌区; g) 闭合系统宜有“打开迹象”; h) 灭菌包裹材料通常使用指示胶带闭合,并根据灭菌方法选择不同种类的胶带。有不同种类的胶带可用于纺织或无纺布的包裹材料。不宜使用挤压包装或医疗器械的方式进行闭合(例如,使用绳索、绳子、松紧带、回形针、钉书钉或类似物品); i) 可重复使用织物作为灭菌包裹材料时,还需要在每次使用前确保灭菌包裹材料的额外适应性要求(见GB/T 19633.1-2015,5.1.11和5.1.12中的要求)。 4.3.2.6.4 可重复使用容器 可重复使用硬质容器用于放置医疗器械及其附件,并在无外部包裹情况下进行灭菌。这种容器通常包括带提手的底部或底座,盖子通过锁定装置固定在底座上,可能还有放置医疗器械的提篮或托盘。该容器包括排气和灭菌介质渗透装置。在不同标准中,被称为“硬质容器”或“可重复使用容器”。 器械盒、器械篮筐或整理托盘属于容纳类设备,但不是无菌屏障系统。宜被包含在无菌屏障系统中。 使用硬质容器时,宜考虑以下方面(见GB/T 19633.1-2015, 5.1.10中的要求)。 a) 仅使用经证明与特定容器、特定的灭菌过程兼容且能够保持无菌的过滤器。过滤器制造商需提供可证明具有此类功能的书面证据; b) 根据制造商说明检查并准备容器; c) 适用于灭菌过程的“打开迹象”指示系统符合容器制造商的要求,并显示无菌屏障系统在预期使用前未被故意或意外开启,以免内容物受到潜在污染; d) 每个容器都需有明显的识别标签和/或信息卡,除非容器设计用于紧急灭菌。识别标签和/或信息卡需适用于灭菌过程; e) 每次使用时检查底座和盖子的密封面是否破损,保证容器适当封闭; f) 器械整理托盘的尺寸适合于特定容器和灭菌方法; g) 需有每次使用后的容器清洁、灭菌和维护过程程序,并得到确认。不得使用超出制造商规定使用年限的容器(见GB/T 19633.1-2015, 5.1.12中的要求)。建立程序,以确保不超出制造商规定使用年限(见GB/T 19633.1-2015, 5.1.12中的要求); h) 与所有无菌屏障系统一样,为了保证无菌取用,容器外侧以及顶部与底部的连接处不得与无菌内容物接触。 4.3.2.7 保护性包装 保护性包装可以在应对环境挑战或多次装卸时保护或适当延长包装和灭菌物品的有效期。特别是,无菌屏障系统的运输或移动可能需要使用保护性包装,以确保运输和搬运不影响无菌屏障系统。需尽量减少搬运无菌包装。无菌屏障系统的完整性与事件相关,与时间无关,这就是为什么防止无菌屏障系统损坏如此重要的原因。 使用保护性包装前,无菌屏障系统需清晰可辨。保护性包装旨在提供额外的保护,以防止损坏和外部因素的不利影响。若在蒸汽灭菌后使用任何保护性包装,则需在彻底冷却干燥后使用。 国家或相关地区法规可能要求使用保护性包装,以避免对手术环境造成潜在污染。这些法规还可能要求在无菌屏障系统进入手术环境前需移除保护性包装。 4.3.3 包装系统性能试验(GB/T 19633.1-2015,5.3,5.4,5.5,6.3) 医疗机构首次使用任何包装系统前,宜进行性能试验,以验证无菌屏障系统或包装系统在灭菌前后满足预期处理、流通和运输严苛条件的能力。无菌屏障系统宜保持其完整性,不会因施加的应力而出现任何孔洞、撕裂或密封/封口破裂。 性能测试宜: 1) 装配无菌屏障系统,其中包含对无菌屏障系统形成最大挑战的医疗器械配置(例如,最大、最重、最密集、最尖锐物品,见GB/T 19633.1-2015, 6.3.4); 2) 宜根据国家或地区监控灭菌效果的要求,准备用于验证测试的样本,以监控灭菌过程的有效性。如通过使用热电偶或数据记录器测量和记录物理参数,生物、化学指示剂或过程挑战装置(PCD)来监控灭菌过程。适应性的确认可以与灭菌过程确认同时进行。宜根据医疗器械和预成型无菌屏障系统制造商的说明对医疗器械进行包装和灭菌; 3) 无菌屏障系统的预期灭菌过程宜考虑混合装载或满载; 4) 无菌屏障系统的流通/搬运/贮存/开启。 宜考虑贮存无菌屏障系统或包装系统的环境和其他条件。将产品紧紧地塞入箱子和存储位置,会增加两组已包装的医疗器械之间发生剪切作用的机会,这可能导致穿孔和撕裂。 许多灭菌站点不与使用地点相邻,宜考虑所有贮存和流通条件。 性能测试后,医疗机构宜目视检验无菌屏障系统的包装完整性(无穿孔或撕裂)和密封完整性,然后确认是否达到灭菌参数。 若需更深入测试,可选择的试验方法见GB/T 19633.1-2015附录B。 若 无 菌 屏 障 系 统 设 计 为 可 重 复 使 用 , 且 制 造 商 预 期 其 性 能 特 性 会 发 生 降 解 ( 见 GB/T 19633.1-2015,5.1.11和5.1.12),则使用的监测或检查系统需清楚的识别制造商提供的使用寿命终点。 4.3.4 无菌屏障系统稳定性评估(有效期)(GB/T 19633.1-2015,6.4) 通常由预成型无菌屏障系统制造商评估无菌屏障系统材料或预成型无菌屏障系统随时间变化保持其性能特性和密封完整性的能力(见GB/T 19633.1-2015, 6.4.7中的要求)。 即使材料已被证明具有可接受的微生物屏障,医疗机构也宜证明组装的无菌屏障系统或包装系统可以在预期环境条件下保持完整性,直到使用时为止。 无菌屏障系统完整性失效与事件有关,与时间无关,取决于无菌屏障系统或包装系统的性能、医疗器械与所选无菌屏障系统之间的相互作用、贮存条件、运输条件和搬运次数。为保证合适的存储环境,医疗机构应考虑多种因素。例如,防止损坏,保持温度和湿度稳定性,避免暴露于灰尘和阳光下,使用保护性包装,尽可能减少搬运次数,将清洁物品和受污染的物品分开放置等。 适当的库存控制和管理系统可以降低无菌屏障系统损坏风险,最大限度地保持包装完整性。 4.3.5 文件资料 医疗机构宜按照计划或标准来评估无菌屏障系统。对比测试结果与可接受准则,并记录评估结果。 宜根据医疗机构的要求保留确认文件和收集的数据。GB/T 19633.1-2015, 7.1要求记录所测试材料的类型、规格、等级和批号、灭菌过程、已知有效期或建议贮存条件、任何对处置或使用的限定、可重复使用材料保养的频次和方式。 4.4 GB/T 19633.2 符合性指南,成型、密封和装配过程确认的要求 4.4.1 总则 GB/T 19633.2讨论了所有包装过程的确认要求。包括装配、填充及以下过程: ——密封过程:组合袋、卷材或纸袋的成型和密封; ——包裹过程:灭菌包裹折叠和灭菌包裹闭合; ——容器封装过程:可重复使用容器闭合。 过程确认依赖于先前安装鉴定(IQ)和运行鉴定(OQ)的数据。这些数据可用于确定关键参数的公差。 宜在正式质量管理体系框架内执行包装过程。医疗机构质量体系之间存在区域差异,关键要素包括(但不限于)有效的文件控制程序、培训控制程序、过程控制/监控程序以及维护(和不断改进)保证包装过程有效性的纠正/预防措施程序。 在GB/T 19633.2中,IQ、OQ和性能鉴定(PQ)的定义均涉及密封或闭合过程中使用的设备。但是,所有成型、密封和装配过程都需要人工操作。因此,人员的执行功能需作为确认的一部分。 通常仅在要安装设备时执行IQ。或者对于仅涉及人员及其执行任务的过程,某些机构可能将标准操作程序(SOP)的开发及其培训视为IQ。OQ和PQ操作员的培训宜记录在这些报告中。 4.4.2 确认方法 4.4.2.1 总则 宜有确认方法或确认的标准程序并形成文件,包括: a) 编制确认计划(见4.4.2.2); b) 实施确认(见4.4.2.3),包括: 1) IQ(见GB/T 19633.2-2015, 5.2); 2) OQ(见GB/T 19633.2-2015, 5.3); 3) PQ(见GB/T 19633.2-2015, 5.4)。 c) 解决故障及采取纠正措施的程序; d) 确认批准(见4.4.2.4); e) 过程控制和日常监控(见4.4.2.5); f) 过程/包装变更和再确认(见4.4.2.6)。 4.4.2.2 编制确认计划 确认计划至少包括以下信息: 可使用附录D.2至附录D.4的确认计划清单。宜针对不同的灭菌方式和无菌屏障系统和/或包装系统组合使用单独的确认计划(制造商、类型等),附录D中表D.1可用于管理目标。 4.4.2.3 确认实施 编制确认计划后,确认活动将根据确认计划执行。4.4.1所述三个过程的具体指南,见以下章节:密封过程(4.4.2.7),包裹过程(4.4.2.8)和容器封装过程(4.4.2.9)。 4.4.2.4 确认批准 记录和评估确认报告宜具有可追溯性,并由已批准确认计划中的负责人批准(见附录D.2至附录D.4)。 偏差宜在确认报告批准前解决并批准。宜评估偏差对确认研究的影响,以确定是否再次研究。 在执行OQ前,IQ报告需得到批准。同时,在执行PQ前,OQ报告宜得到批准。 完成每个步骤后,宜研究失效或偏差情况,在开始下一个确认步骤前确定根本原因并采取纠正措施。宜评估是否需要完全或部分重复之前的确认步骤。宜使用正式系统管理纠正性或预防性措施,并评估和记录措施的有效性。 4.4.2.5 过程控制和日常监控 建立控制包装过程的程序,并在日常操作中保持在既定参数范围内。 日常监控并记录关键过程参数。 4.4.2.6 过程/包装变更和再确认 注:影响已确认过程状态以及再确认需求的变更清单如下: ——无菌屏障系统原材料改变; ——新设备; ——过程和/或设备从一个地点或机构移向另一地点或机构; ——灭菌过程改变; ——审查最终用户投诉或不合格产品,质量或过程控制显示有下降的趋势; ——超出最初评估的最坏情况下参数范围的无菌屏障系统内容物变化; ——更改运输路径或方式(例如,从建筑物内部到建筑物之间的运输,这可能会对包装产生挑战)。 可通过再确认表明操作人员具备有效实施过程所需的知识和能力,还可以用于重新培训人员和重新调整实践。 4.4.2.7 预成型无菌屏障系统密封过程确认(组合袋、卷材或纸袋成型及密封) 4.4.2.7.1 总则 预成型无菌屏障系统制造商需提供预成型密封的密封过程确认信息,此信息可用于医疗机构封口确认。 4.4.2.7.2 安装鉴定 正确安装密封设备。密封设备宜由有校准资质的机构提前校准,且医疗机构宜有持续校准计划以保证密封界面的密封参数正确。此外,还需对用户进行操作密封设备的培训。 IQ考虑以下方面:清洁度、温度、湿度等环境条件;记录/操作培训;操作手册或程序。 宜解决以下问题: 4.4.2.7.3 运行鉴定 医疗机构使用的密封温度范围宜由预成型无菌屏障系统制造商和密封设备制造商提供的信息确定。 预成型无菌屏障系统制造商通常提供规定压力和时间条件下的温度上限和下限。 密封设备制造商通常提供设备监控关键参数的信息。 接触压力和密封/停滞时间的参数范围通常由密封设备制造商提供,宜确保设备能够获得预成型无菌屏障系统制造商推荐的限值。在使用条件下,不同无菌屏障系统可能需要不同的密封温度。 根据提供的信息,在上限和下限参数密封无菌屏障系统,并评估所密封产品的质量。在使用前检查密封设备是否已校准。 培训操作人员,并评估其进行热密封过程的能力。 包装应按照程序文件进行装配。宜考虑最坏情况配置(见附录H)。 在各变量上限和下限参数下生产样品,并进行评估。对于已成功确认的极限值,所有样本都宜满足可接受准则。无菌屏障系统密封的可接受准则包括: a) 规定密封宽度下密封完整; b) 无通道或密封开口; c) 无刺破或撕裂; d) 没有贯穿密封宽度的褶皱或折痕; e) 在经过预期灭菌过程后,封口剥离处无影响无菌取用的材料分层或纤维撕裂。 使用破坏性试验评估密封时,宜为每个密封参数准备多组包装样品。 为了在灭菌后达到上述可接受准则,可能需要确定最小密封强度(例如,YY/T 0698.5规定蒸汽灭菌过程的最小参考值为1.5 N/15mm,其他灭菌过程的最小参考值为1.2 N/15mm)。若在OQ期间未进行密封强度测试,PQ期间不满足可接受准则的风险较高,可能需要再次执行OQ。 使用合适的试验系统(例如,使用染料渗透试验套件或其他密封完整性指示器,见附录A.7.3)检查上述质量特性并形成文件。 注1:密封完整性指示卡所用材料宜与组合袋或卷材(例如,YY/T 0698.3)的透气材料相同。若在上限和下限参数下均满足质量特性要求,则设定值通常是这两个值的平均值(例如,下限= 170℃,上限= 190℃,则密封温度= 180℃)。 注2:附录F.1中的OQ清单可用于确定密封温度。 4.4.2.7.4 性能鉴定 PQ宜证实该过程(包括设备和操作人员)在规定操作条件下能持续生产出满足可接受准则的无菌屏障系统。 考虑以下方面: a) 无菌屏障系统的评估宜在无菌屏障系统密封和灭菌后进行; b) 确认过程中使用的批次信息是确认记录的一部分。批次信息包括但不限于: c) 使用制造商推荐的程序检查测试设备和密封设备的校准。宜在密封前进行; d) 样品宜密封并评估。所有样品均宜满足可接受准则。样本量指南见4.4.2.2 h);为了在灭菌后达到可接受准则,可能需要确定最小密封强度(例如,YY/T 0698.5规定蒸汽灭菌过程的最小参考值为1.5 N/15mm,其他灭菌过程的最小参考值为1.2 N/15mm)。使用破坏性试验评估密封时,宜为每个密封参数准备多组包装样品。 e) 生产三批或三组无菌屏障系统,宜包括潜在重要变化来源,例如操作人员、时间、材料(数量、来源、批次)、无菌屏障系统内容物,宜包括代表最大挑战(最坏情况)的包装内容物; f) 无菌屏障系统的测试样品宜使用预先被确认的、能够证明无菌屏障系统适用性的灭菌过程进行灭菌。三批试验样品宜在三个单独循环中通过相同灭菌过程,证明其可重复性; g) 无菌屏障系统应在经过灭菌工艺和预期的最苛刻的处理、流通和贮存条件之后,采用OQ制定的可接受准则进行评估,并记录结果,见4.3.3和GB/T 19633.1-2015, 6.3。 注:使用附录G.1中的PQ清单进行记录。 4.4.2.7.5 自封袋或胶带封合袋 当有热封设备和组合袋时,不鼓励使用自封袋或胶带封合袋。若使用,则宜确认其装配和封口过程(见GB/T 19633.2-2015, 5.1.1中的要求)。GB/T 19633.2-2015第5章中所有适当的要素和步骤应予以说明。 4.4.2.8 包裹过程确认(灭菌包裹的折叠和闭合) 4.4.2.8.1 安装鉴定 包裹过程通常是手动过程,IQ宜考虑以下方面:环境条件(如,清洁度、温度、湿度);记录/操作培训;操作手册或程序。 4.4.2.8.2 运行鉴定 宜有包装装配的文件程序。该方法宜考虑4.3.2.3和4.3.2.6.3。包裹组装和闭合指南可以从包装制造商处获取。 对操作人员进行能力培训并评估。折叠方法需形成阻碍微生物通过的曲折路径(见附录C)。 按程序进行包装装配。在装配时,宜包括最坏的配置情况,见附录H。样品宜被闭合/密封并评估。 所有样品都宜满足可接受准则。样本量指南见4.4.2.2 h)和附录L。 根据评估闭合的试验方法,每个批次准备多组样本。 评估无菌屏障系统的完整性和有效闭合。可接受准则宜包括但不仅限于: a) 闭合连续性和完整性; b) 无通道、开口或间隙; c) 无穿孔或撕裂; d) 开启时无材料分层或分离; e) 开启包装需证明无菌屏障系统能够实现内容物的无菌取用; f) 灭菌参数满足要求; g) 干燥参数满足要求。 除了评估闭合的无菌屏障系统外,包装还宜被打开并评估是否符合所记录的装配程序。 注:可使用附录F. 2中的OQ清单。 4.4.2.8.3 性能鉴定 PQ需证实在规定操作条件下,包裹过程能持续生产出满足可接受准则的无菌屏障系统。 1) 操作人员; 2) 时间和日期; 3) 灭菌过程、参数和循环次数; 4) 无菌屏障系统材料; 5) 闭合胶带; 6) 无菌屏障系统内容物。 4.4.2.9 容器封装过程确认(可重复使用容器的填充和闭合) 4.4.2.9.1 总则 GB/T 19633.2和本指南讨论了可重复使用容器的填充和闭合,但未讨论在重复使用前,对这些容器的清洁或净化方式。在医疗机构使用过程中,可重复使用容器在填充和封口前宜进行经确认过的清洗/净化过程。 可重复使用容器进行过程确认时,需保证每个容器(例如,垫片状态、容器等)满足制造商的所有要求,并形成记录。 4.4.2.9.2 安装鉴定 容器填充和闭合通常是手动过程,IQ宜考虑以下方面:环境条件(例如,清洁度、温度、湿度);记录/操作培训;操作手册或程序。如使用设备,则宜根据GB/T 19633.2-2015, 5.2进行IQ。 4.4.2.9.3 运行鉴定 宜有评估容器损坏、填充和闭合的程序,并形成文件,见4.4.2.3和4.4.2.6.4的要求。可从制造商处获取详细信息。 对操作人员进行能力培训并评估。在运行鉴定前,必须编写SOP并获得批准。 宜根据制造商说明和医疗机构的文件程序对容器进行清洁、检查、装载,并使用“打开迹象”指示系统闭合。确定或选择容器的内容物时,宜包括内容物的最坏情况配置(例如,容器装载的重量、体积和材料)。操作人员进行的无菌屏障系统典型调整(例如,更换过滤器等)也应被包括。所有样本宜满足可接受准则。样本量指南见4.4.2.2 h)。根据不同使用条件,可使用相同容器一次或多次装配样本用于鉴定。根据评估闭合的试验方法,每个批次准备多组样本。 评估无菌屏障系统的完整性和有效闭合。可接受准则包括但不仅限于: a) 密封、容器咬合面、容器系统边缘和盖子,确保无凹陷或碎裂; b) 过滤器固定架和紧固件(例如,螺钉和铆钉)牢固,无扭曲或磨损; c) 锁闭装置运作正常; d) 过滤介质的完整性不受影响; e) 垫片柔韧,牢固固定,无断裂或切口; f) 阀门工作正常; g) 闭合连续性和完整性; h) 过滤器、阀门及灭菌介质口未损坏; i) 能够在不损坏内容物的情况下打开容器; j) 容器允许内容物无菌取用; k) “打开迹象”指示系统有效且完整; l) 灭菌参数满足要求; m) 干燥参数满足要求。 除了评估闭合的无菌屏障系统外,容器还宜被打开并评估是否符合装配程序中的清洁、检查和装载要求。 注:可以使用附录F.3所述OQ清单。 4.4.2.9.4 性能鉴定 容器制造商宜提供证据证明容器在指定灭菌过程中的适用性及其保持内容物无菌性的能力。PQ证明在特定操作条件下,装载、填充和闭合过程将持续生产出满足可接受准则的无菌屏障系统。 无菌屏障系统的评估宜在无菌屏障系统闭合和灭菌后进行。宜考虑以下几点: a) 确认过程中使用的批次信息是确认记录的一部分。批次信息包括但不限于: b) 根据医疗机构的文件程序要求装配闭合三个批次或三组无菌屏障系统,宜包括潜在重要变化来源,例如操作人员、时间、材料(数量、来源、批次)、无菌屏障系统内容物,宜包括代表最大挑战(最坏情况)的包装内容物。若在几个不同灭菌过程中使用相同无菌屏障系统,则宜对每个灭菌过程进行确认; c) 所有样品宜满足可接受准则,样本量指南见4.4.2.2 h); d) 无菌屏障系统应在经过灭菌工艺和预期的最苛刻的处理、流通和贮存条件之后,采用OQ制定的可接受准则进行评估。在蒸汽灭菌时,除了OQ制定的可接受准则外,还应在灭菌后对内容物进行评估,确保内容物充分干燥; e) 三批试验样品宜在三个单独循环中通过相同灭菌过程,证明其可重复性; f) 记录所有评估结果; 注:可以使用附录 G.3 所述包装过程 PQ 清单进行记录。 g) 若发现包装失效,则宜通过调查确认根本原因,见附录R。 4.5 质量体系 GB/T 19633.1-2015, 4.2和GB/T 19633.2-2015, 4.1要求正式的质量体系,但未提供进一步指南。 5 工业指南 5.1 通用指南 5.1.1 质量体系 特定设计要求的开发的更多信息见 5.3 和附录 J 有关设计输入的规定。 5.1.2 试验方法 GB/T 19633.1 和 GB/T 19633.2 要求对证明符合这些标准的所有试验方法进行确认(见附录 O)。关于 GB/T 19633.1 和 GB/T 19633.2 所述试验方法的要求指南见本文件附录 A。 最好采用经过系统地、跨实验室研究的试验方法,因其重复性、再现性及在某些情况下的灵敏度均已进行了研究。在特定实验室执行这些试验方法时,必须证明其准确性和重复性至少达到实验室间研究的再现性。 可以使用独立开发或科学文献所述的试验方法。但是,必须确定试验方法是否满足所需灵敏度,并且准确性和重复性满足预期接受准则。 5.1.3 取样 取样计划宜适用于包装系统,反映风险承受能力,并以统计有效原理为基础(GB/T 19633.1-2015,4.3)。更多信息见 5.8.2 和附录 I.4。 5.2 设计输入 考虑材料和/或包装系统设计前,宜建立一套设计输入(GB/T 19633.1-2015, 6.2.2 和 6.2.3)。这些设计输入将用于评估材料和/或设计。 设计输入将反映用户需求。该信息将来自于用户工程、生产、营销、法规需求等。设计输入实例包括医疗器械属性、医疗器械保护要求、销售单元配置、灭菌过程、配送、装卸和使用环境(GB/T19633.1-2015,6.1 和 6.2)。 注:设计输入的开发指南见附录 J。 5.3 材料选择和评估 5.3.1 总则 本部分相关信息见附录 A。 5.3.2 灭菌要求指南(GB/T 19633.1-2015,5.1.6 e)和 5.3) 评估对医疗器械、过程和最终用途十分重要的材料特性时,必须注意适合灭菌过程的材料特性(例如,用于气体灭菌的透气性)以及对严苛灭菌过程的承受力。更多灭菌信息见附录 B。 5.3.3 安全要求指南(GB/T 19633.1-2015,5.1.5 和 5.1.6) 无菌屏障系统选择材料时,宜满足基本的安全要求。宜了解并控制材料来源、历史和可追溯性。宜评估化学特性。这通常包括毒性以及材料与医疗器械之间可能发生的化学相互作用评估。一般还包括测试是否存在有毒重金属。更多信息见附录 A. 5.3.4 屏障要求指南(GB/T 19633.1-2015,5.1.4 和 5.1.6) 医疗器械和选定灭菌方法的屏障要求有助于确定适当的无菌屏障和包装系统材料。可以使用各种方法评估材料特性,并评估保护医疗器械所需的屏障或屏障水平。需要考虑的各个方面,包括透气性、微生物屏障、空气(氧气)、水分、温度和透光性等。详见附录 A 的屏障部分。 5.3.5 可视性和外观要求指南 可视性和外观要求(如适用)由外观美学(例如,高光泽度或磨砂效果的无菌屏障系统)、标识方法(例如,需要高清晰度无菌屏障系统的标签内插页)以及器械可视性(或遮盖)需求决定。宜考虑雾度、光泽度、不透明度和透明度等方面。更多信息见附录 A。 5.3.6 物理特性要求指南(GB/T 19633.1-2015,5.1.6 c),5.1.7 e)和 6.3.2) 无菌屏障系统宜能够在使用前保护医疗器械的无菌性、功效和/或功能。无菌屏障系统或预成型无菌屏障系统的物理特性要求取决于内容物的重量和结构轮廓、保护性包装的类型(如适用)、贮存条件和配送系统要求。有几个因素会影响材料性能,例如抗穿刺性、耐磨性、抗撕裂性、抗揉搓性、厚度和基本重量。这些因素详见附录 A。评估物理特性的试验方法仅说明材料的特性,但不能直接预测无菌屏障系统的性能,通常需要通过实验室模拟包装系统性能试验评估特定医疗器械无菌屏障系统的性能。 5.3.7 热密封性要求指南(GB/T 19633.1-2015,5.1.6 d)和 5.1.8 c)) 宜测试封口的拉伸剥离强度,并检查确定结果是否满足无菌屏障系统所需的密封强度。其它接受准则包括可剥离性、目力检测封口外观和打开过程中材料失效。通常通过密封拉伸测试选择材料组合,并评估灭菌前后的热密封效果。可以使用各种不同的密封强度试验方法。见 GB/T 19633.1 附录 B。YY/T0681.2 详细介绍了部分密封强度试验方法,还包括有关技术差异影响的信息。 5.3.8 加工要求指南(GB/T 19633.1-2015,5.1.2 至 5.1.9) 评估包装材料与设备和加工条件的兼容性时,宜考虑确保一致可靠生产无菌屏障系统预定要求。常用于评估过程要求和条件的方法是尺寸测量、摩擦系数(COF)、可密封性和涂层重量,详见附录 A。 5.3.9 印刷要求指南(GB/T 19633.1-2015,5.4) 设计印刷时必须注意包装材料的特性。字体或字号可能与特定基材不适宜,也宜考虑印刷颜色、图稿排版和稿件等因素。新的基材评估宜考虑可印刷性。印刷油墨的属性宜防止物理和化学降解,以及预期暴露灭菌过程的过程指示物发挥功能。还宜考虑成型前/后的印刷效果,防止图形外观的严重变形,使文字信息难以辨认。更多关于特定印刷要求的材料评估信息见附录 A。 5.3.10 清洁度及微粒要求指南(GB/T 19633.1:2015, 5.1.7d)) 一般清洁度要求是包装材料没有污渍、灰尘、油渍和其他污染。微粒可以是嵌入或松散的异物或母材碎片。更多关于微粒的信息见附录 A。固有颗粒物水平取决于选择的包装材料。通常使用标准尘埃图(GB/T 1541)或其他适用标准估计微粒污染程度。 5.3.11 器械-包装系统相互作用 材料硬度、黏着度、变色或其他属性可能与医疗器械的使用相互作用。 无菌屏障系统的组成可以迁移至医疗器械(可沥滤物),并与其内容物相互作用,导致不利影响。同样,医疗器械材料可以迁移至包装系统材料中,从而产生不利影响。宜评估风险,并进行适当的适应性研究。 5.4 无菌屏障系统和保护性包装设计(包装系统开发) 5.4.1 关键设计要素 应有形成文件的无菌屏障系统和保护性包装设计与开发程序(GB/T 19633.1-2015, 6.2.1)。包装系统设计过程的一个关键组成部分是收集和评估设计输入(GB/T 19633.1-2015,6.2.2 和 6.2.3),见 5.3 和附录 J。 包装功能开发宜包含在设计控制系统或过程中。宜从医疗器械整个开发周期的早期开始设计最终灭菌医疗器械包装系统。参与医疗器械开发过程并深刻理解医疗器械的所有属性是非常重要的,包括相关医疗器械、灭菌和制造规范。 可以与合同包装商共同设计无菌屏障系统和保护性包装(即包装系统)。合同包装商使用指南见附录 P。 5.4.2 包装系统设计步骤 5.4.2.1 无菌屏障系统设计 a) 预成型托盘和盖材(GB/T 19633.1-2015,A.3.2); b) 预成型组合袋(GB/T 19633.1-2015,A.3.3); c) 预成型灭菌纸袋(GB/T 19633.1-2015,A.3.4); d) 预成型顶头袋(GB/T 19633.1-2015,A.3.5); e) 可重复使用容器(GB/T 19633.1-2015,A.3.12); f) 由折转路径形成闭合的灭菌包裹(GB/T 19633.1-2015,A.3.11); g) 形成无菌屏障系统并完成密封的包装–成型/装入/密封和四边密封(GB/T 19633.1-2015,A.3.6和 A.3.7)。 注:确保供应商能力和使用设备的公差是合理的。 5.4.2.2 保护性包装设计 保护性包装对无菌屏障系统及其内容物提供物理保护。 根据质量体系程序要求,规定并记录保护性包装的材料、尺寸、几何形状和物理特性。 5.4.2.3 包装系统样品 a) 若评估通过,则继续进行可行性测试; b) 若包装系统样品不通过,则返回至设计阶段。 注:包装系统设计可行性测试指南见附录 S.2。 5.4.2.4 包装系统的标签设计注意事项 5.5 包装过程可行性评估 5.5.1 无菌屏障系统的生产过程 确定无菌屏障系统的生产过程。建立生产过程图或流程图。明确制造、装载、密封和包装过程的每个步骤。在每个步骤中,分析无菌屏障系统或包装系统的潜在失效风险,以及可能导致质量问题的变化来源。评估风险,由此确定控制和重新设计包装过程的方式(指南见附录 K)。指明包装系统材料和医疗器械移动状况。关于详细分析的指南见附录 S。关于确定过程参数的指南见附录 Q。 5.5.2 设备安装鉴定指南 确定工艺图或流程图中所定义的各阶段/设备的设备 IQ 要求。IQ 相关讨论参见 GB/T 19633.2-20155.2 和本文件 5.8。 a) 使用现有设备,确定是否使用新工具或当前工具,并进行 IQ 评估; b) 若使用新设备,运行 IQ。 5.5.3 打样或试运行 建议为最初性能测试生产样品,降低在包装过程确认过程中的失效风险。 无菌屏障系统性能测试样品需使用相关工艺参数生产。 5.6 无菌屏障系统设计可行性评估 5.6.1 一般注意事项 a) 无菌屏障系统是否已装入医疗器械并保护其免受物理损坏; b) 无菌屏障系统及内容物之间的相互作用。 5.6.2 无菌屏障系统试验方法 5.6.3 最坏情况可行性条件 注:关于最坏情况的指南见附录 H。 在与包装系统相关的众多因素中,确定最坏情况,或者在此过程中被认为的最坏情况,以更好地确认其可行性。这些因素包括但不仅限于: a) 无菌屏障系统生产(密封参数等); b) 灭菌过程(参数、循环次数等):初步可行性测试时,可能无需对样品进行灭菌; c) 运输配置:需要了解医疗器械运送至客户的方式; d) 配送环境:需要了解医疗器械运送至客户的方式; e) 可行性评估时通常无需有效期试验;但可开展加速老化试验。加速老化试验是通过将完成所有过程的保护性包装系统置于升高温度的受控环境下,模拟时间对包装系统的影响。通常根据Arrhenius 反应速率函数所述动力学,通过假设包装材料降解以估计实时时间。如需进一步指南见 GB/T 19633.1-2015,附录 B。 5.6.4 包装系统的合格/不合格状态 5.7 无菌屏障系统生产过程确认 5.7.1 总则 无菌屏障系统确认通常需要医疗器械制造商和/或合同包装商及包装供应商的其他职能部门参与。对于预成型无菌屏障系统,医疗器械包装商的过程通常局限于无菌屏障系统填充及密封。若采用成型-装入-密封方法,医疗器械包装商通常需要确认无菌屏障系统的成型以及医疗器械装入及密封。可结合GB/T 19633.2 及本指南一起使用。 5.7.2 过程确认方案开发 5.7.3 方案中详述的确认活动执行情况 5.7.3.1 安装鉴定(若不满意先前 IQ 工作)(GB/T 19633.2-2015,5.2) IQ 活动可以包括: a) 确立安装清单: b) 验证设备是否在设计参数范围内运行; c) 进行软件确认; d) 建立环境条件; e) 建立设备操作 SOP 并得到批准,培训操作员并保持记录; f) 验证关键过程参数是否受到控制和监视,如: g) 建立校准程序和时间表; h) 建立预防性维护清洁程序和时间表。 5.7.3.2 运行鉴定(GB/T 19633.2-2015,5.3) 注:最坏情况的更多指南见附录 H。 5.7.3.3 性能鉴定(GB/T 19633.2-2015, 5.4) 通常宜评估三个成功的批次。三个批次生产运行在正常操作条件下完成,并且可被其他生产过程间隔。三次 PQ 生产运行都需要成功,期间不得有失败运行。宜考虑足够的运行时间,以及转换、中断和多班次的影响。验证生产的无菌屏障系统是否符合方案中的可接受准则。 5.7.4 确认结果评估 评估确认结果(GB/T 19633.2-2015,5.4.7)。评估 5.7.3 的相关活动,验证是否符合 5.7.2.4 和 5.7.2.5所述的可接受准则。宜记录与方案的任何偏差,并通过审查和评估确定是否满足验证方案。 5.7.5 过程确认批准 过程确认宜由相关人员审批(GB/T 19633.2-2015,5.5)。 5.7.6 持续过程控制和监视的记录 建立持续过程控制和监视的记录(GB/T 19633.2-2015,5.6)。通常包括: a) 监视和记录关键过程参数; b) 按照质量体系要求对无菌屏障系统进行过程测试。 注:选择的监视设施必须适用于过程监视。根据数据和过程知识选择适用于质量体系所述过程的监视设施。 5.8 包装系统设计确认 5.8.1 概述 包装系统设计确认(GB/T 19633.1-2015,6.3, 6.4)包括: a) 建立确认方案,即包装系统测试计划; b) 执行方案中的状态调节和测试; c) 评估得到的测试结果是否符合方案中的可接受准则; d) 过程确认的正式批准。 5.8.2 确认方案 确认方案包括以下详细信息: ——待测试包装系统使用的无菌屏障系统生产需在已确认或将确认过程中生产。试验样品应是生产或等同于已确认过程的生产; ——无菌屏障系统或包装系统应装有预期的医疗器械/包装配置和使用经确认的灭菌过程进行灭菌。若有规定,可以经历多次灭菌过程以模拟最坏情况; ——待测试包装系统需包括装有医疗器械或医疗器械替代品的无菌屏障系统、标签、使用说明书(IFU)和其他包装层。 5.8.4 记录确认结果 测试结果与方案所述的可接受准则进行对比,并记录方案合格/不合格评估。 根据质量体系要求保存确认报告和收集的数据。最终报告获得批准。 5.9 再确认 附 录 A (资料性) 包装材料和无菌屏障系统的选择、评估和测试——工业和医疗机构指南 A.1 概述 谨慎选择与可灭菌的医疗器械相适应的包装材料(GB/T 19633.1-2015,5.1,第 6 条)。无菌屏障系统要考虑的方面包括灭菌的适应性、运输和处理相关的稳健性、屏障特性,以及与最终使用医疗器械相关的各种注意事项。最好在医疗器械设计过程的早期就进行包装系统材料的选择,若直到设计过程快结束前才进行,可能延误医疗器械上市,影响有效期或其他最终用途。医疗包装制造商在这个过程中扮演了重要角色。 试验方法是评估无菌屏障系统和/或包装系统和包装材料的适用性及监控其生产过程的有效手段。最好使用有精度和偏差说明的试验方法,见 GB/T 19633.1-2015 附录 B。选择哪种试验方法取决于医疗器械在其整个生命周期中的要求,所检测的性能参数需与医疗器械无菌屏障系统和/或包装系统属性密切相关。并非所有试验方法始终都适用。此外,有些试验方法适合研发,有些试验方法更适合监控。附录 A 并不作为可选试验方法的概要,而是要作为对较常见方法及其背后原理的简要概述。相关指南见YY/T 0698 系列或 YY/T 1759。 A.2 与灭菌过程的适应性 A.3 安全性考虑因素 其他安全相关问题是随着时间推移,可提取物可能从包装材料析出,可能污染医疗器械或环境。可根据 ASTM D4754 进行可提取物测试。 A.4 屏障指南 A.5 医疗器械的可视性和外观 可以通过几个标准试验方法对比包装材料,估计材料满足医疗器械可视性或外观目标的能力。雾度是指光通过材料时的散射,可以根据 GB 2410 进行测试。光泽度是指基材的反射率或表面光泽,可以根据 ASTM D2457 测试。不透明度是指材料阻止光传输的能力,更多信息见 ASTM D589。 A.6 材料物理特性 除了经受灭菌过程外,医疗器械的无菌屏障系统和/或包装系统还需根据需要保持医疗器械的无菌性和有效性。医疗器械的形状和质量、保护性包装的类型(如适用)以及运输和贮存系统都对无菌屏障系统和/或包装系统的物理特性形成了挑战。实际使用情况是确定医疗器械适应性的唯一方法,标准物理特性提供了在特定应用中选择潜在材料的依据。只有几个特性(若有)表示了特定医疗器械挑战无菌屏障系统和/或包装系统的方式。包装材料制造商将提供这些特性的数值,可用作筛选工具,通常作为典型值来提供,而不是有严格公差的规范值。 ——医疗器械和无菌屏障系统; ——无菌屏障系统和无菌屏障系统; ——无菌屏障系统和保护性包装。 由于目前没有预测这些影响的试验,通常需要对真实医疗器械进行无菌屏障系统和/或包装系统性能试验。 A.7 无菌屏障系统完整性 A.8 密封强度和胀破强度 A.9 材料加工指南 A.10 印刷指南 A.10.1 初次使用的包装材料可能需要对其印刷适应性进行评估。材料的印刷适应性与其可湿性或表面张力有关。表面张力的测量可以利用接触角仪器或达因笔,从而得知材料表面张力等级或者材料的可印刷面。一些被处理的材料表面经过一段时间后会发生衰弱,从而影响其印刷适性。 A.10.2 油墨在材料表面的附着力会影响印刷外观和清晰度,也会影响包装材料的涂层功能。由于每个应用对于附着力的要求不同,因此必须由包装材料的生产商或者使用者来制定可接受标准。参见 YY/T0681.7。 a) 印刷本身就是为了传递信息,因此不得有漏印、污点、污迹或过底,导致印刷内容不正确或难以辨认(GB/T 19633.1-2015,5.4)。在热封区的印刷可能影响材料的热封性,同样热封过程也可能影响油墨和/或印刷清晰度。其他考虑因素可能包括与所选灭菌过程的兼容性,因温度和/或耐化学性可能影响印刷; b)在运转过程中,已印刷的包装材料因磨损造成的印刷字体难以辨认会改变无菌屏障系统和/包装系统的图文外观。在实验室条件下,通过对比表面印刷材料的耐磨性与设定标准,可以粗略估计运输和装卸过程中的影响。如需进一步指南见 ASTM D5264; c) 无菌屏障系统和/或包装系统材料的印刷表面可能会在其生命周期中接触化学品。化学品会降解、软化、污染和消除印刷图文,影响其外观和易读性。宜评估材料对已知或预期化学品的相对耐受性。如需进一步指南见 YY/T 0681.6。 A.11 清洁度和微粒 A.12 加速老化和环境挑战 a) 加速老化——加速老化是一种模拟时间对包装影响的技术,通过在代表实际贮存条件的受控环境中使产品/包装系统经受高温。等效时间通常通过假设包装材料退化符合 Arrhenius 反应速率函数所述动力学来估计,更多讨论见 YY/T 0681.1; b) 环境挑战——环境挑战是指包装经受极端温度和/或湿度和/或其他环境条件的过程,目的是确定包装对环境压力的敏感性。与加速老化相反,环境挑战通常包括温度和湿度相等或超过包装生命周期内可能遇到的温度和湿度条件或变化,或者两者皆有。更多指南见 YY/T 0681.16。 实时老化温度选择和加速老化时长计算实例,见 YY/T 0681.1。 不同材料对温度和湿度的敏感性不同,对于特定材料作为无菌屏障系统和/或包装系统部件的适用性结论可能无效。如纸张对相对湿度特别敏感。过度干燥会对强度性能产生负面影响,过高湿度会导致霉菌生长。有些复合材料可能在极端条件下失效,尽管实时或控制老化很少或从未出现这样的结果。 Arrhenius 方程一般用于确定高温对均相反应速率的影响。简单来说,使用 Q10 = 2 计算,假设每升温 10 °C,老化过程将加倍。如在 55 °C 条件下老化 45 天相当于在 25 °C 条件下老化一年。(更多讨论见 AAMI TIR17 和 YY/T 0681.1) 宜谨慎选择温度,防止材料因实际不会发生或者超出其推荐使用范围的条件而损坏。加速老化过程中的湿度影响指南见 YY/T 0681.1。 宜谨慎选择加速老化测试的温度,防止引起任何材料转变或无菌屏障系统和/或包装系统变形,或者发生非线性变化,如结晶、产生自由基和过氧化物降解。 考虑湿度在加速老化中的作用时,必须认识到相对湿度是指在该温度条件下,空气中相对于容量的悬浮水量。因此,在加速老化研究期间保持与环境水平相同的相对湿度百分比有着固有危险,必然会导致更高的水分暴露(高于实时老化期间的水分暴露)。与此相关的详细讨论,见参考文献。 宜根据对产品分布和使用条件的了解仔细选择温度和湿度水平。除了线性问题外,上述温度和湿度讨论也与环境挑战有关。常用配送系统的方法见 YY/T 0681.16。 附 录 B (资料性) 灭菌注意事项——工业和医疗机构指南 B.1 概述 a) 材料选择——透气或不透气材料; b) 气体灭菌法——一般需要透气无菌屏障系统; c) 辐照灭菌法可使用不透气或透气无菌屏障系统。有时可使用无菌屏障系统减少辐照期间出现的异味; d) 适应性——承受灭菌过程的能力: B.2 环氧乙烷灭菌 ——GB 18279.1; ——GB/T 16886.7; ——GB/T 18279.2; ——YY/T 1302.1; ——YY/T 1302.2; ——AAMI TIR19; ——AAMI TIR20; ——YY/T 1268。 B.3 伽马辐照灭菌 a) 医疗器械无菌屏障系统和/或包装系统和油墨需要使用能够承受电离辐照的材料。交联作用和断链作用会导致外观和功能性物理特性变化,或者使某些材料发生变性。可以更换为辐照耐受材料; b) 辐照灭菌方法可能使温度升高,宜加以考虑; c) 包装物密度是一个重要因素,宜在灭菌过程中保持一致来维持剂量测定放行的安全; d) 若医疗器械存在无法通过气体灭菌的区域或者需要不透气无菌屏障系统和/或包装系统,则宜考虑这种灭菌方法; e) 可优化包装物尺寸,与辐照器机架的尺寸相适应。 a) 时间:灭菌时间相对较短,但是类似密度的医疗器械同时灭菌,可能会延长灭菌时间。整个过程使用剂量测定放行,无需在灭菌后等待; b) 装载量宜基于辐照容器中使用的托盘尺寸以及剂量分布研究。 ——GB 18280.1; ——GB 18280.2; ——GB/T 18280.3; ——YY/T 0884; ——YY/T 1613; —— GB/T 31995; ——YY/T 1608。 B.4 电子束灭菌 a) 选择能够承受电子束辐照的医疗器械、无菌屏障系统和油墨等的材料。与伽马辐照相比,电子束辐照对材料的影响相对较小; b) 包装物密度是一个重要因素,宜在灭菌过程中保持一致来维持剂量测定放行的安全; c) 若医疗器械存在无法通过气体灭菌的区域,电子束灭菌是一个很好的选择。 a) 时间:无菌屏障系统通过传送带运输,有时候需要通过两次。由于这个过程使用剂量测定放行,所以无需在灭菌后等待; b) 宜控制医疗器械/无菌屏障系统和/或包装系统的朝向,使医疗器械各部位都能够接触到电子束,即尽量避免“遮蔽”等问题; c) 装载量:受到密度要求和设备/传送带特性限制; d) 若电子束灭菌整合至生产线中,是一种有效的灭菌方法。 a) GB/T 16841; b) 伽马辐照标准(见附录 B.3.4)。 B.5 X射线灭菌 a) 医疗器械或无菌屏障系统和/或包装系统需使用能够承受电离辐照的材料。X 射线灭菌可以在低剂量条件下短时间暴露,交联作用仍会导致外观(泛黄)和功能性物理特性变化; b) X 射线可以穿透包装材料,进入医疗器械结构,无菌屏障系统和/或包装系统可没有透气材料。 c) 灭菌循环过程不包括真空阶段,对无菌屏障系统封口的压力有限; d) 托盘或手提装载密度是一个重要因素,宜在灭菌过程中保持一致来维持剂量测定放行的安全; e) 优化无菌屏障系统和/或包装系统设计,匹配合同灭菌器托盘或手提材料装卸系统,减少灭菌周期内的无效区域。 a) 效能还取决于医疗器械的无菌屏障系统和/或包装系统、运输包装或和托盘摆放; b) 装载量:基于设备限制,及来自于生产计划和灭菌确认过程输入的确认要求。 B.6 湿热(蒸汽)灭菌 a) 医疗器械、无菌屏障系统和/或包装系统和油墨不宜对水蒸气、冷凝水和高温敏感; b) 无菌屏障系统和/或包装系统宜有允许蒸汽进出的透气区域。蒸汽通过可渗透部分的速度宜能够在真空和/或充气过程中保证无菌屏障系统的完整性。宜确保无菌屏障系统和/或包装系统中的每个无菌屏障系统的摆放不会妨碍其渗透性。避免透气材料与不透气材料紧密接触,以免阻碍蒸汽渗透; c) 医疗器械需要有通道,使蒸汽渗透,并持续接触医疗器械的所有区域。 a) 时间:湿热(蒸汽)灭菌通常不到 2 小时,无需通风。产品需要等待生物指示剂的结果,除非使用参数放行; b) 装载量:受到灭菌设备特性和医疗器械灭菌过程确认限制。 ——GB 18278.1; ——ISO/TS 17665-2。 B.7 湿热不透气包装灭菌 a) 医疗器械、无菌屏障系统和/或包装系统和油墨宜对饱和蒸汽湿度和高温不敏感; b) 无菌屏障系统内部需要有水分,才能产生灭菌所需的蒸汽和压力; c) 医疗器械需要有通道,使蒸汽和/或加压水渗透并持续接触医疗器械的所有区域。 a) 时间:湿热不透气包装灭菌需要几个小时,无需通风。产品需要等待生物指示剂的结果,除非使用参数放行; b) 装载量:受到灭菌设备特性和医疗器械灭菌过程确认限制; c) 可剥离无菌屏障系统灭菌时,通常灭菌过程需要超压控制。 B.8 干热灭菌 医疗器械、无菌屏障系统和/或包装系统和油墨需能承受高温。通常在几个小时内经受 160℃或以上高温,或者在较低温度条件下延长循环时间。 a) 时间:干热灭菌需要几个小时,无需通风。产品需要等待生物指示剂的结果,除非使用参数放行; b) 装载量:受到灭菌设备特性和医疗器械灭菌过程确认限制。 B.9 过氧化氢灭菌 a) 等离子体,也被称为低温过氧化氢气体等离子体(LTHPGP)灭菌; b) 过氧化氢,是与气体等离子体灭菌相似的汽化灭菌过程,但不完全相同。两个灭菌过程都使用汽化过氧化氢,但气体等离子体灭菌还有一个等离子生成步骤。 a) 在已确认循环中,医疗器械、无菌屏障系统和/或包装系统和油墨需能承受 55℃温度和 80%相对湿度; b) 使用等离子体灭菌时,医疗器械、无菌屏障系统和/或包装系统和油墨宜与等离子体和等离子体产生过程相适应; c) 医疗器械,、无菌屏障系统和/或包装系统和油墨需对深度真空或过氧化氢不敏感; d) 医疗器械需要能承受灭菌设备允许的压力变化速率; e) 医疗器械需要有气体进出部分,使气体接触医疗器械的所有区域; f) 无菌屏障系统和/或包装系统需有允许气体进出的透气区域。气体通过可渗透部分的速度宜能够在真空和/或充气过程中保证无菌屏障系统的完整性; g) 纤维素材料不得用于过氧化氢灭菌。 a) 时间:过氧化氢灭菌大约 1 个小时。通常无需通风。某些装载可能需要通风或额外真空/压力清洗,防止气体进入工作场所。产品需要等待生物指示剂的结果,除非使用参数放行; b) 装载量:受到灭菌设备特性和医疗器械灭菌过程确认限制; c) 应通过污染控制装置来处理过氧化氢废气,以避免进入工作场所或环境大气。 B.10 臭氧灭菌 a) 医疗器械和无菌屏障系统和/或包装系统(包括油墨)需能承受高湿度、多次深度真空和臭氧环境。可向医疗器械制造商和灭菌器制造商咨询医疗器械与臭氧的适应性问题; b) 无菌屏障系统和/或包装系统需有允许气体进出的透气区域。气体通过可渗透部分的速度宜能够在真空和/或充气过程中保证无菌屏障系统的完整性; c) 医疗器械需要有气体进出部分,使气体接触医疗器械的所有区域。 a) 时间:臭氧灭菌需要几个小时,无需通风。可使用符合法规要求的化学指示剂和自含式生物指示剂监测灭菌过程。产品需要等待生物指示剂的结果,目前无法使用参数放行; b) 装载量:受到灭菌设备特性和医疗器械灭菌过程确认限制。宜根据灭菌器制造商使用说明书的建议装载。 B.11 二氧化氯(ClO2或CD)灭菌 a) 聚烯烃非织造布和各种透明薄膜、铝箔复合材料和硬质塑料与二氧化氯相适应。有些纸张也可以接受。不得使用未漂白的瓦楞纸箱; b) 医疗器械和无菌屏障系统和/或包装系统(包括油墨)宜能够承受氧化作用和高湿条件(相对湿度通常为 55%-70%)。不同灭菌循环设计的气体浓度(通常为 5 mg/L -30 mg/L)和湿度不同; c) 医疗器械需要有气体进出部分,使气体接触医疗器械的所有区域; d) 无菌屏障系统和/或包装系统需有允许气体进出的透气区域。气体通过可渗透部分的速度宜能够在真空和/或充气过程中保证无菌屏障系统的完整性。 a) 时间:过程步骤通常包括: b) 装载量:受到灭菌设备特性和医疗器械灭菌过程确认限制; c) 效能还取决于构建满载产品的生产时间;满载通常是几个到多个托盘; d) 可以在一定程度上根据医疗器械和微生物要求定制灭菌周期; e) 在灭菌过程中,将二氧化氯浓度降低至安全水平。需要测试医疗器械的灭菌残留物和副产物。 附 录 C (资料性) 包裹方法实例——医疗机构指南 C.1 概述 下图介绍了灭菌前的几种医疗器械包裹方法。这些示例并不是唯一的包裹方法,还有其他可接受的方法。包裹时可以依次进行,也可同时进行。不同包裹层可以采用不同的包裹方法。需注意限制胶带和标签覆盖区域,以确保有足够的多孔区域用于有效灭菌和干燥。包裹方法的接受度取决于要包裹的医疗器械,需由使用者决定。 C.2 信封式包裹方法 包裹步骤如图 C.1 至 C.4 所示。 ' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
图 C.2 信封式包裹方法步骤 4 至步骤 6
图 C.3 信封式双层同时包裹方法
图 C.4 信封式双层依次包裹方法 C.3 平行包装/方形折叠法包裹 平行包装/方形折叠法包裹步骤如图 C.5 至 C.9 所示。 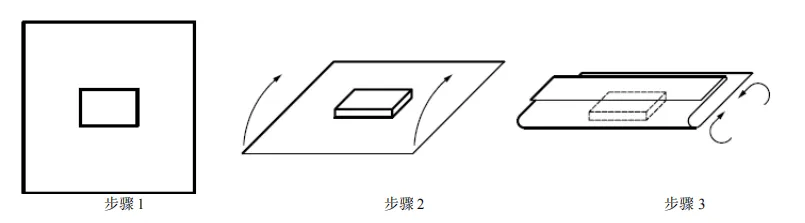 图 C.5 平行包装/方形折叠法包裹 步骤 1 至步骤 3 图 C.6 平行包装/方形折叠法包裹 步骤 4 至步骤 7 图 C.7 平行包装/方形折叠法包裹 步骤 8 和步骤 9 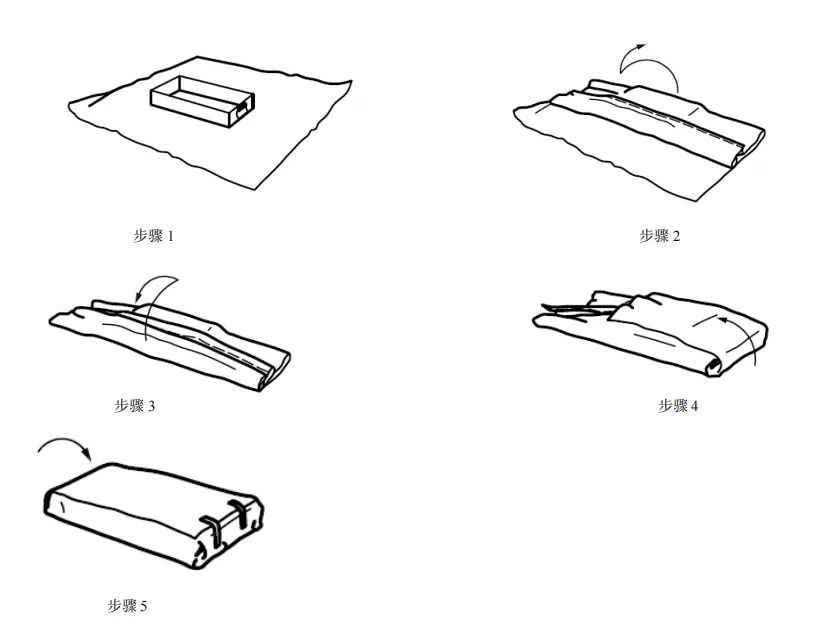 图 C.8 平行包装/方形折叠法包裹双层同时包裹
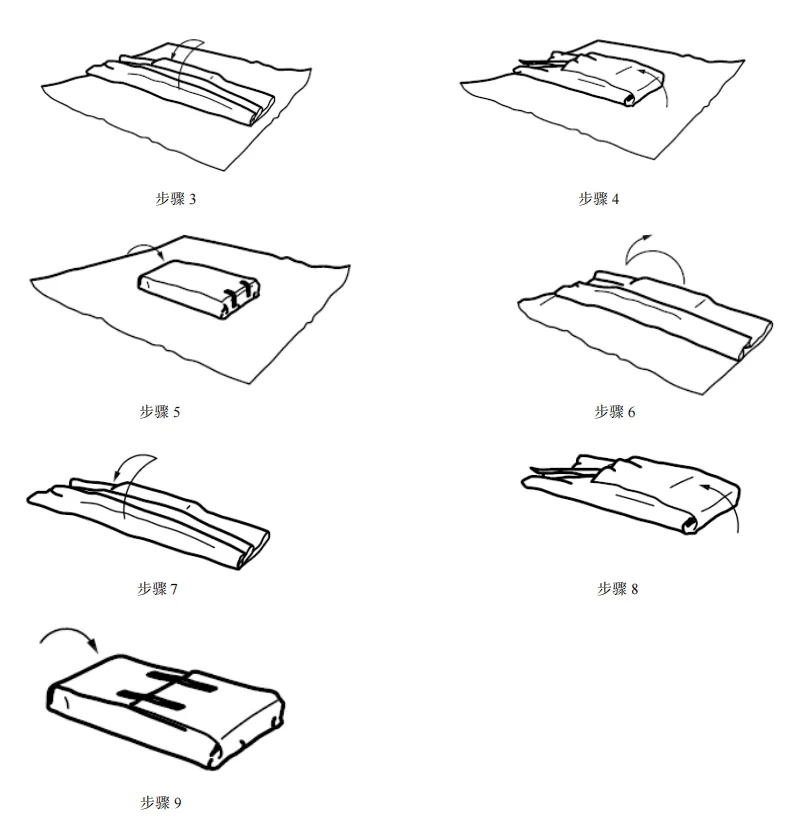 图 C.9 平行包装/方形折叠法包裹双层依次包裹 C.4 巴斯德法或滚压法 巴斯德法或滚压法包裹步骤如图 C.10 所示。
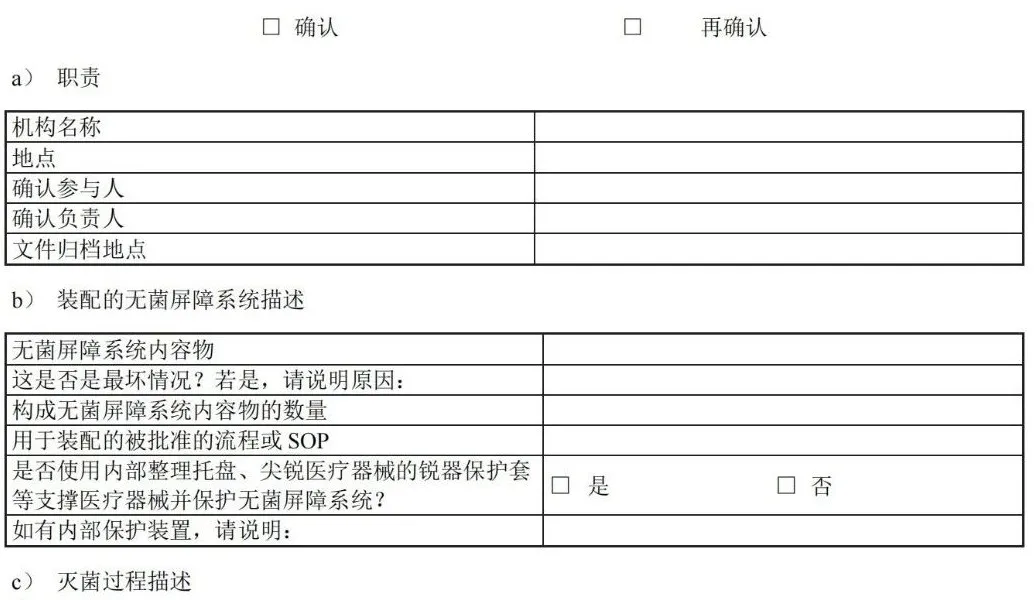
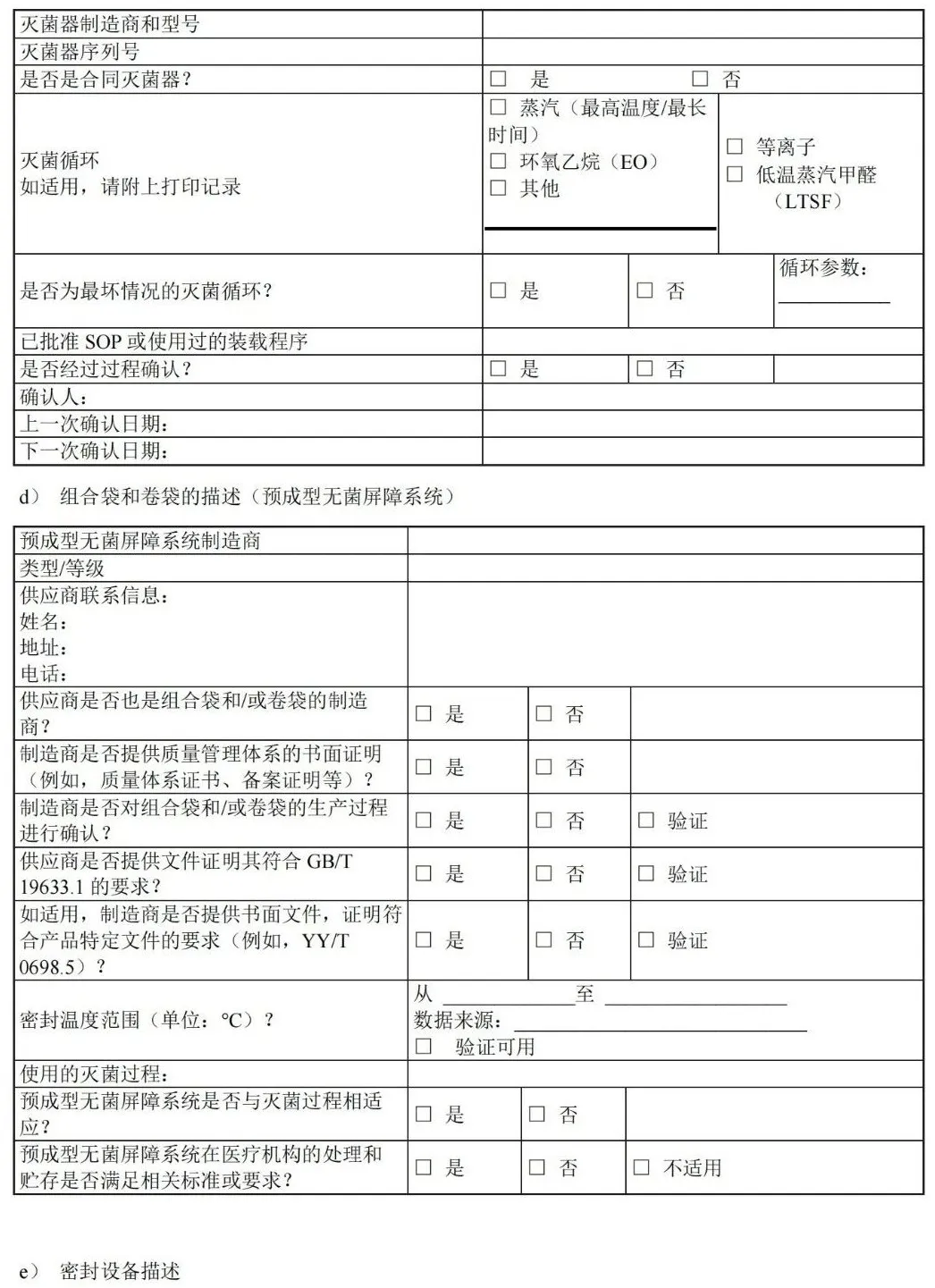
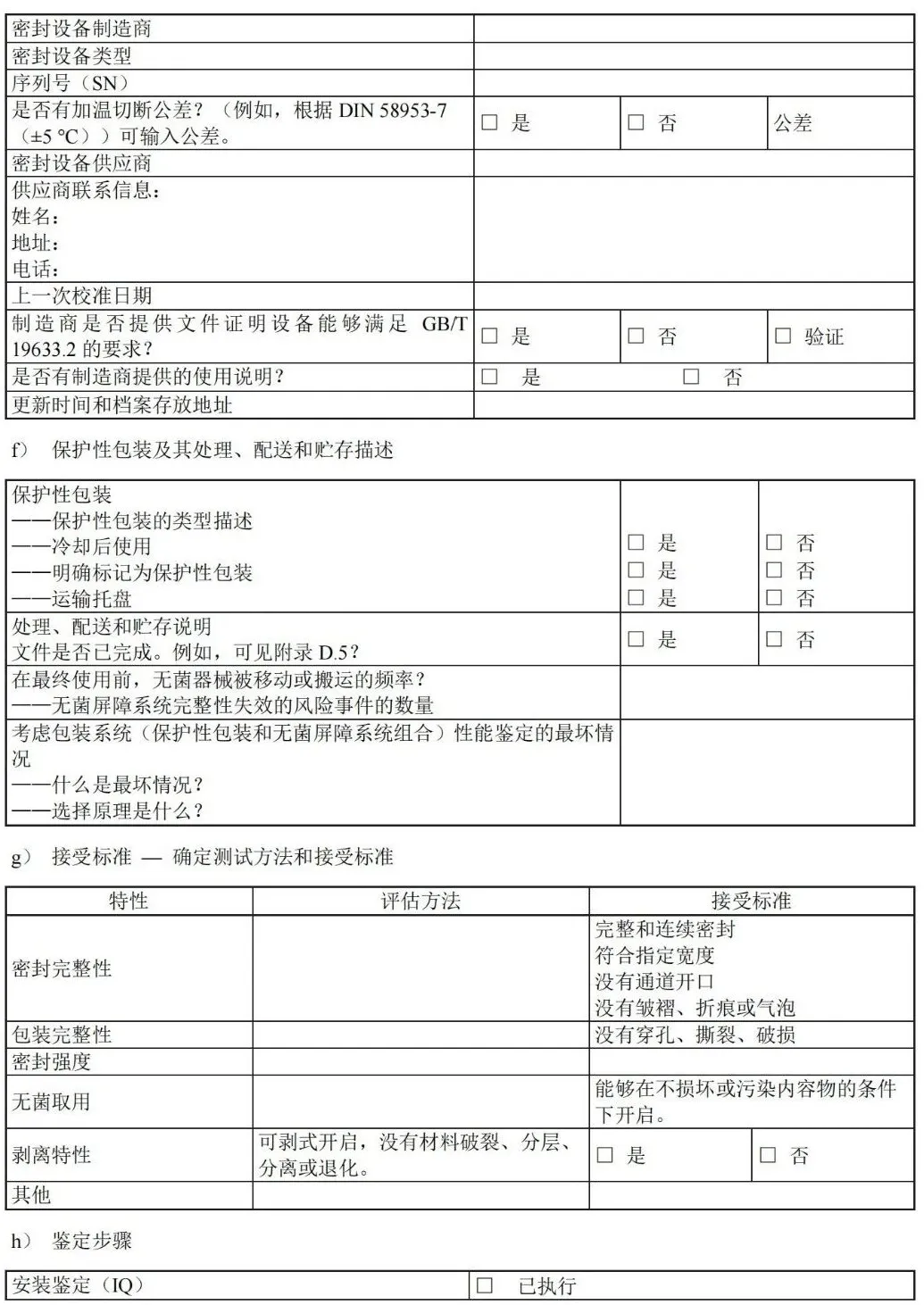
图 C.10 巴斯德法或滚压法 附 录 D (资料性) 确认计划文件——医疗机构指南 D.1 概述 附录 D 中的清单可用于实施并记录包装过程确认。可与灭菌过程确认和/或再确认相结合,并形成文件。下表可用于规划确认的次数。 表 D.1 可用于确定计划执行的过程确认的次数。 D.2 确认计划清单:预成型无菌屏障系统的热密封过程(PSBS:组合袋、卷袋等)
D.3 确认计划清单:灭菌包裹过程
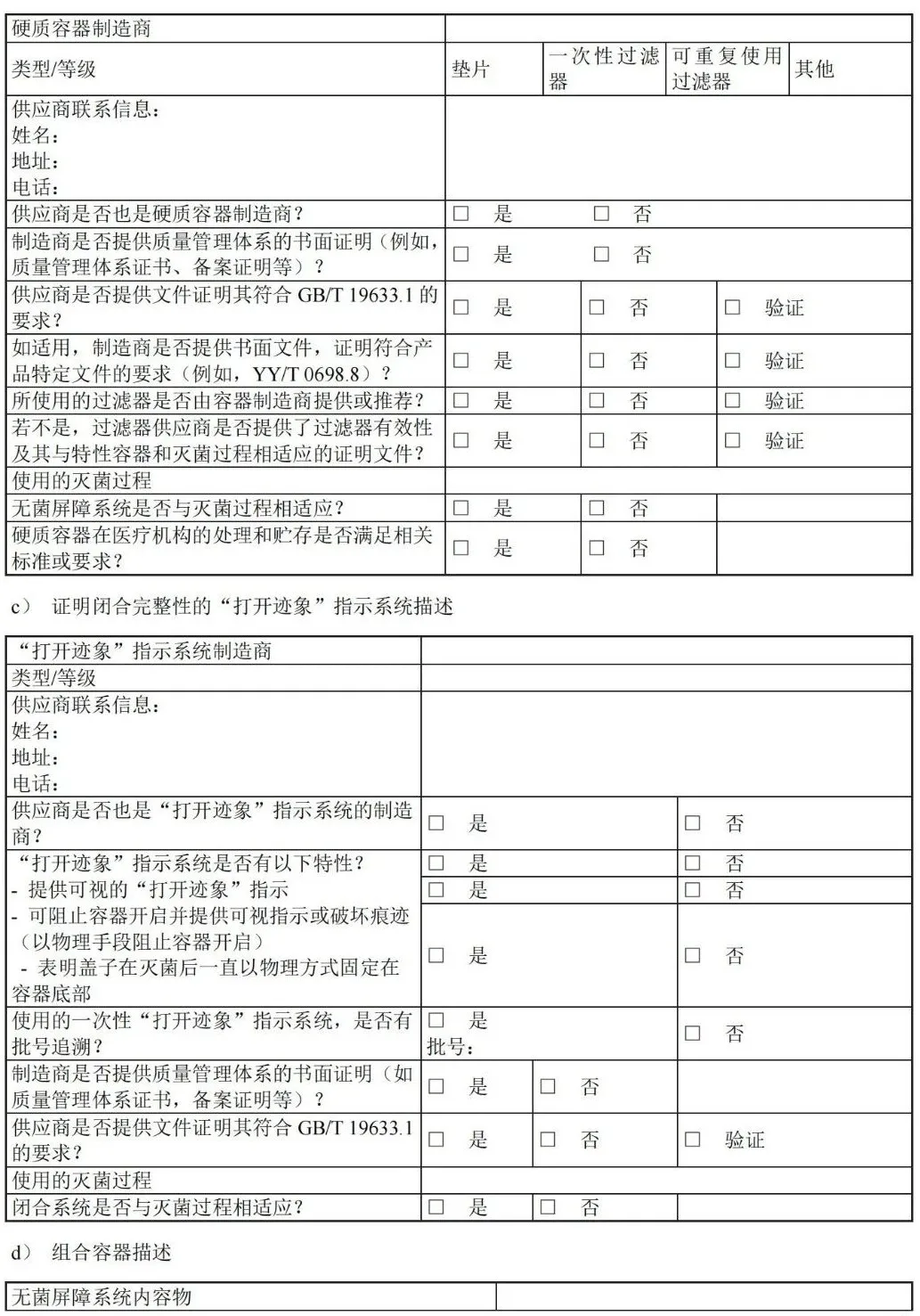
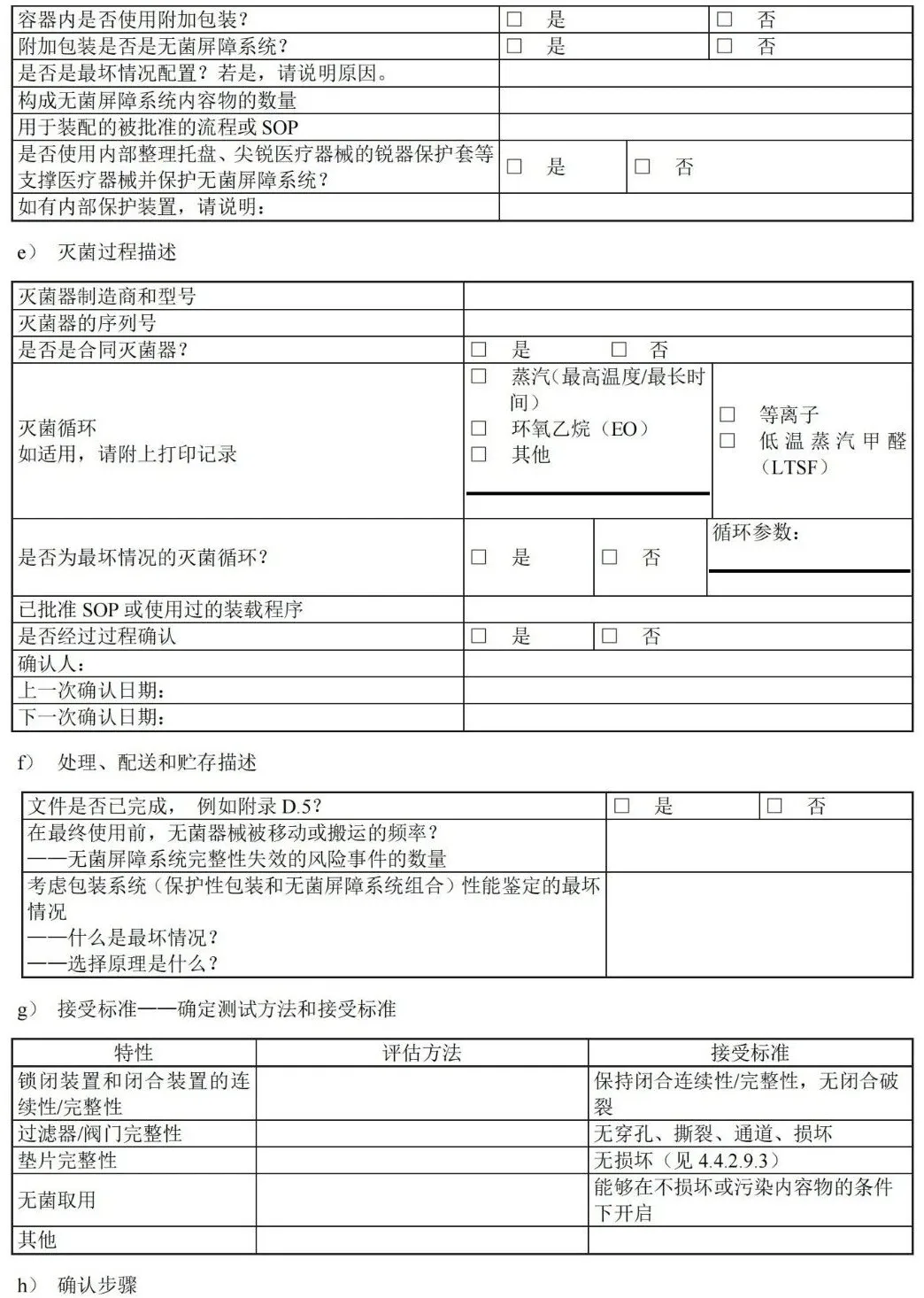
D.4 确认计划清单:容器封装过程
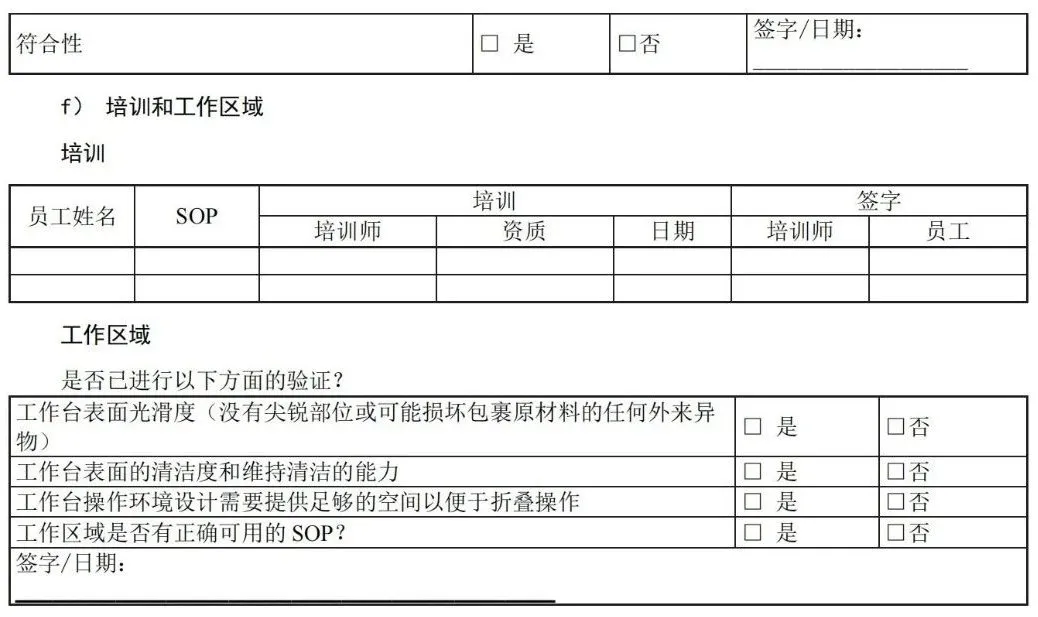
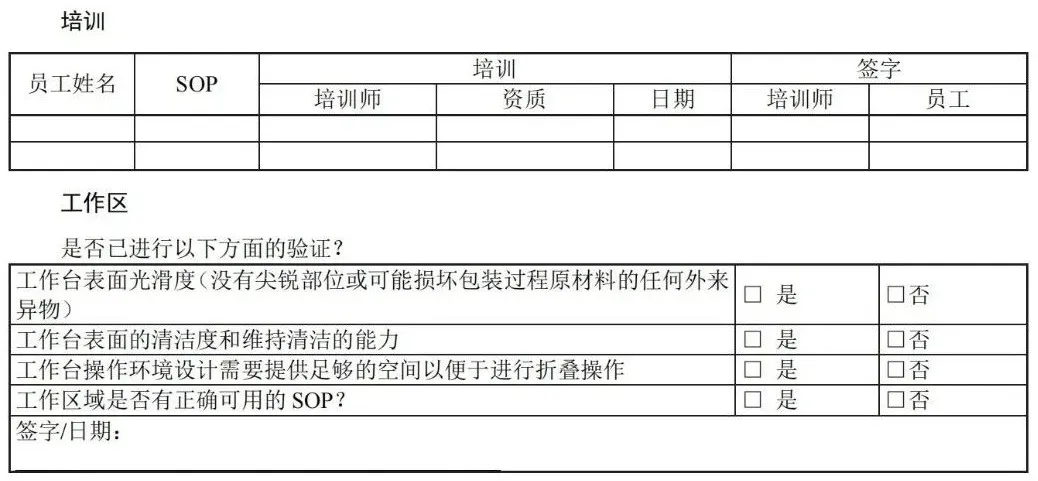
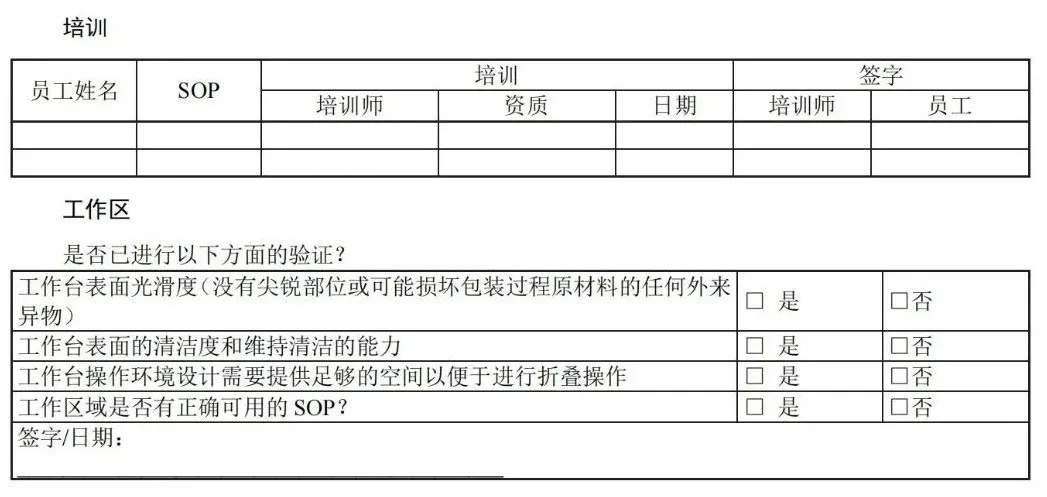
D.5 处理和配送清单:处理、配送和贮存
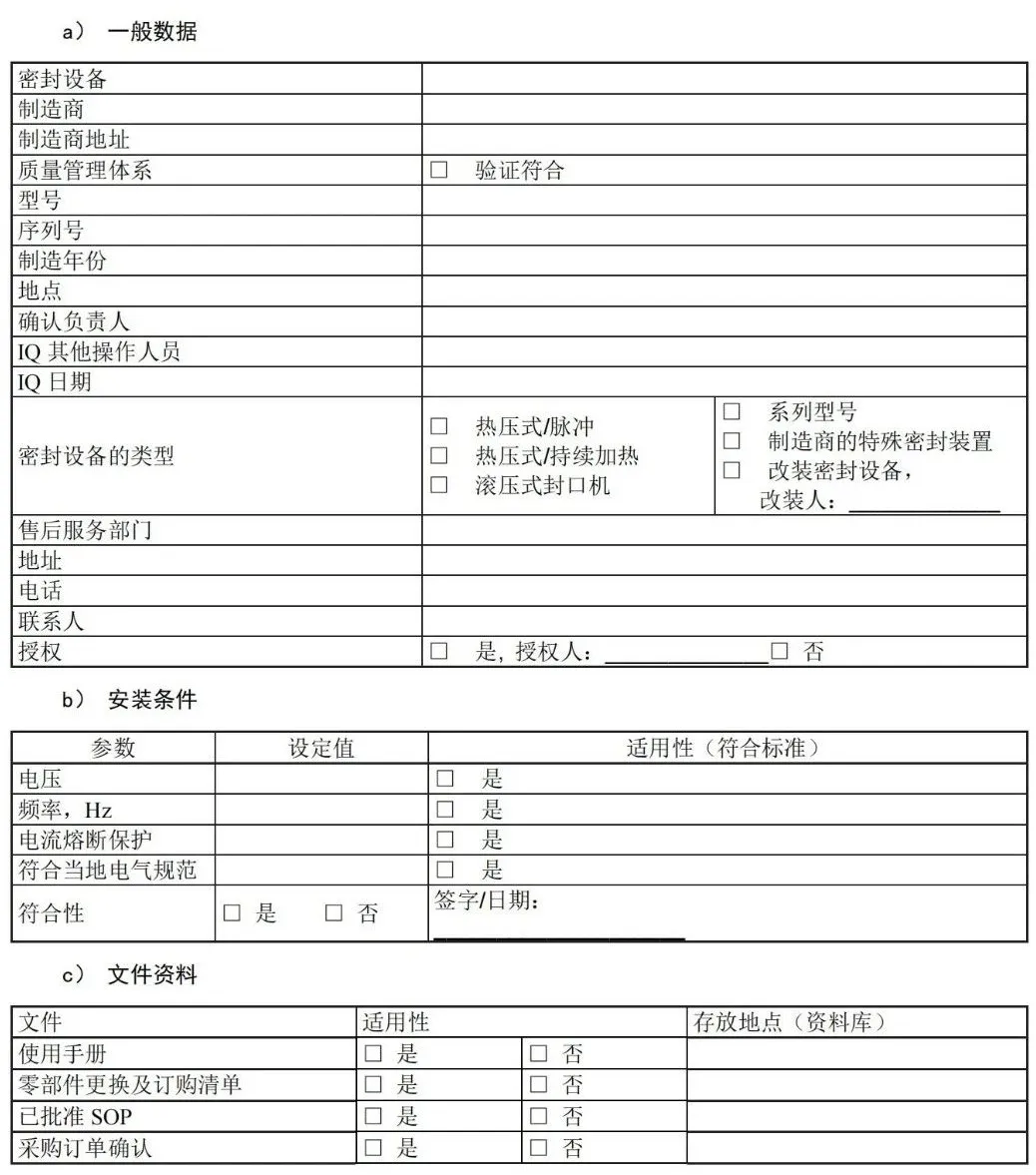
附 录 E (资料性) 安装鉴定文件——医疗机构指南 E.1 安装鉴定(IQ)清单:热密封过程
E.2 安装鉴定(IQ)清单:灭菌包裹过程 通常,只有在进行设备安装时才会执行 IQ。另外,某些机构对于仅涉及人员及其生产的过程,例如 SOP 开发及其培训,也可能会视为 IQ 过程。OQ 和 PQ 过程中对于相关具体操作员的培训也应当记录在报告中。
E.3 安装鉴定(IQ)清单:硬质容器封装过程 通常只有在进行设备安装时才会执行 IQ。另外某些机构对于仅涉及人员及其生产的过程,诸如 SOP开发及其培训,也可能会视为 IQ 过程。OQ 和 PQ 过程中对于相关具体操作员的培训也应当记录在报告中。
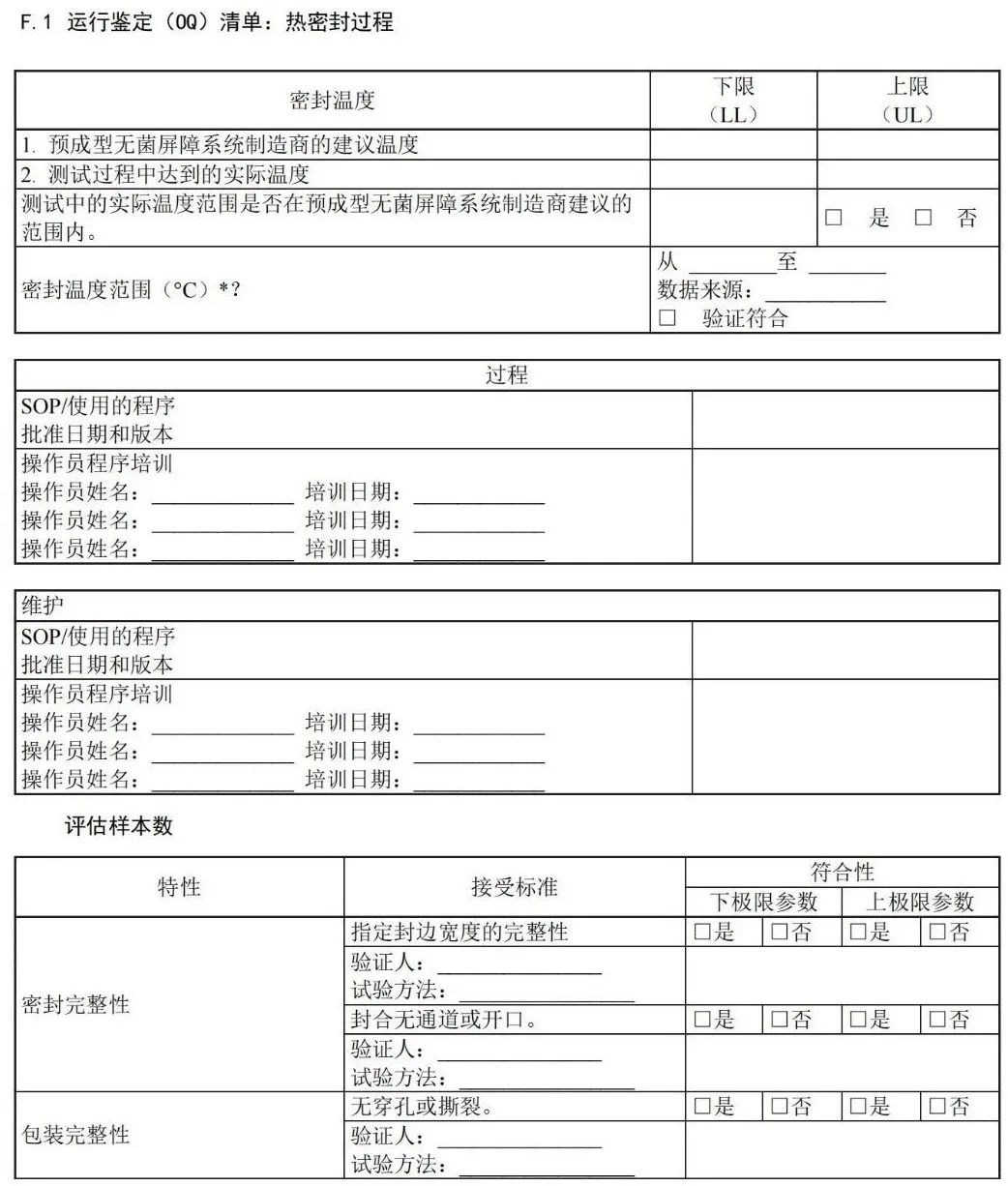
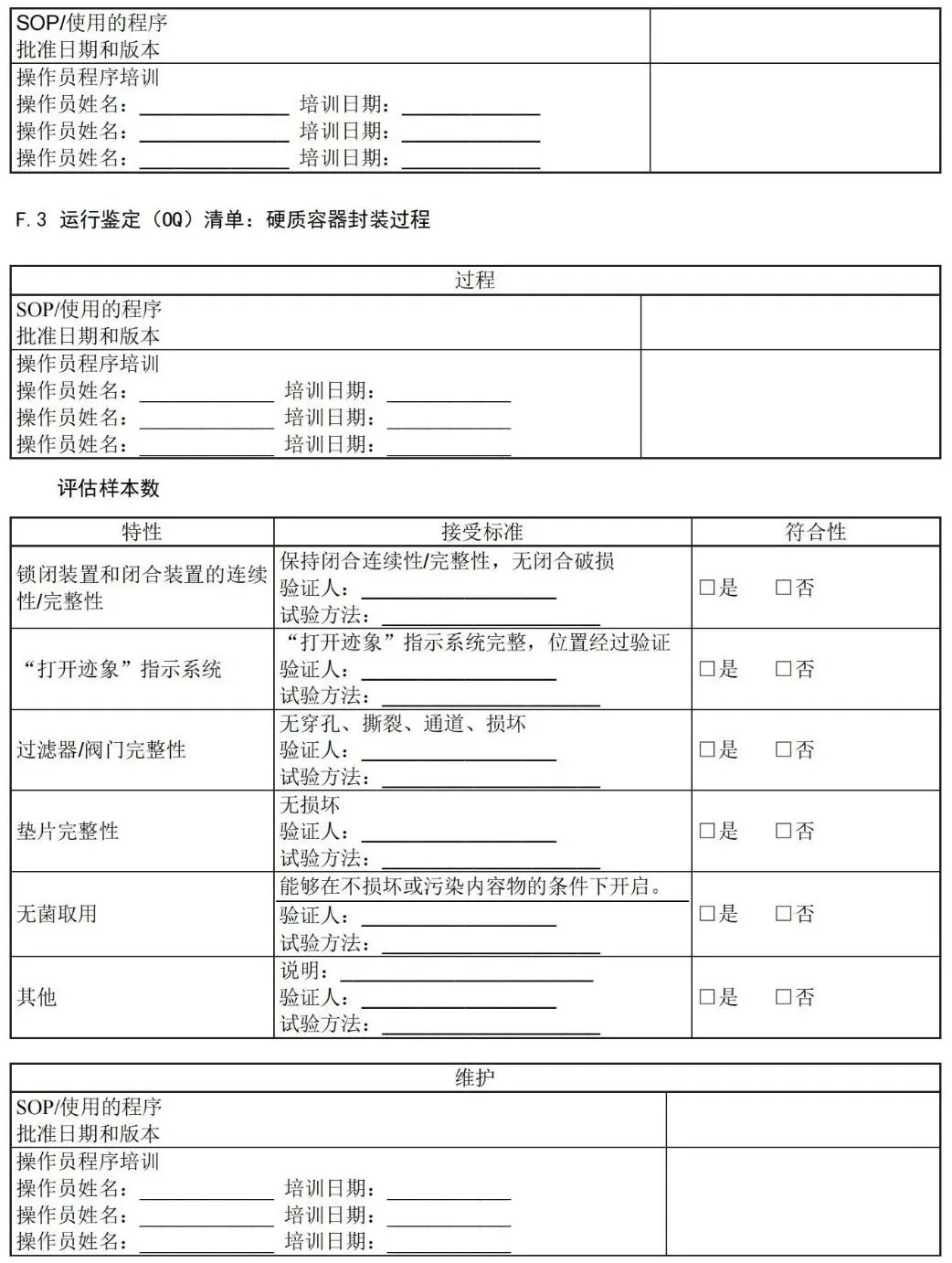
附 录 F (资料性) 运行鉴定文件——医疗机构指南
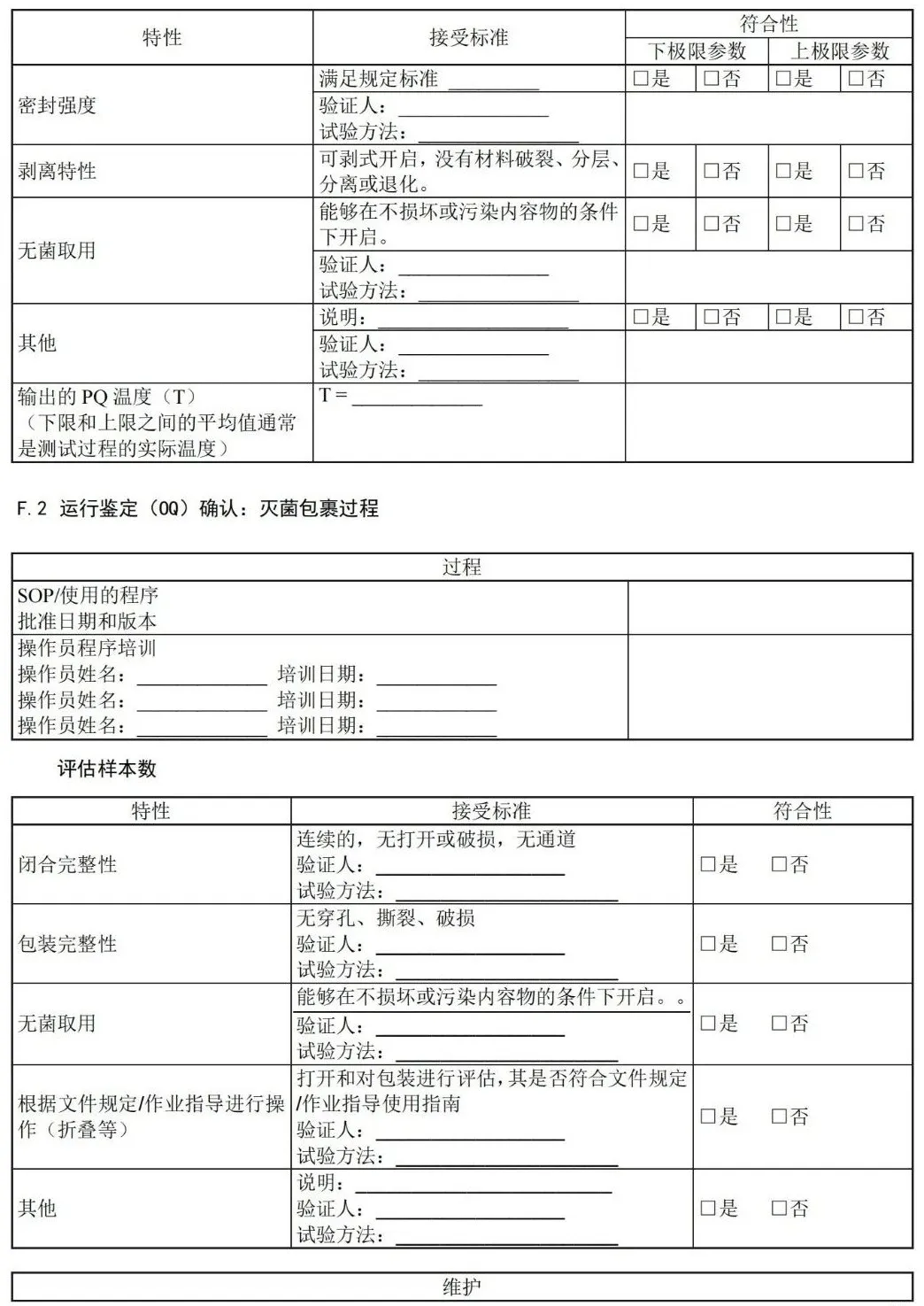
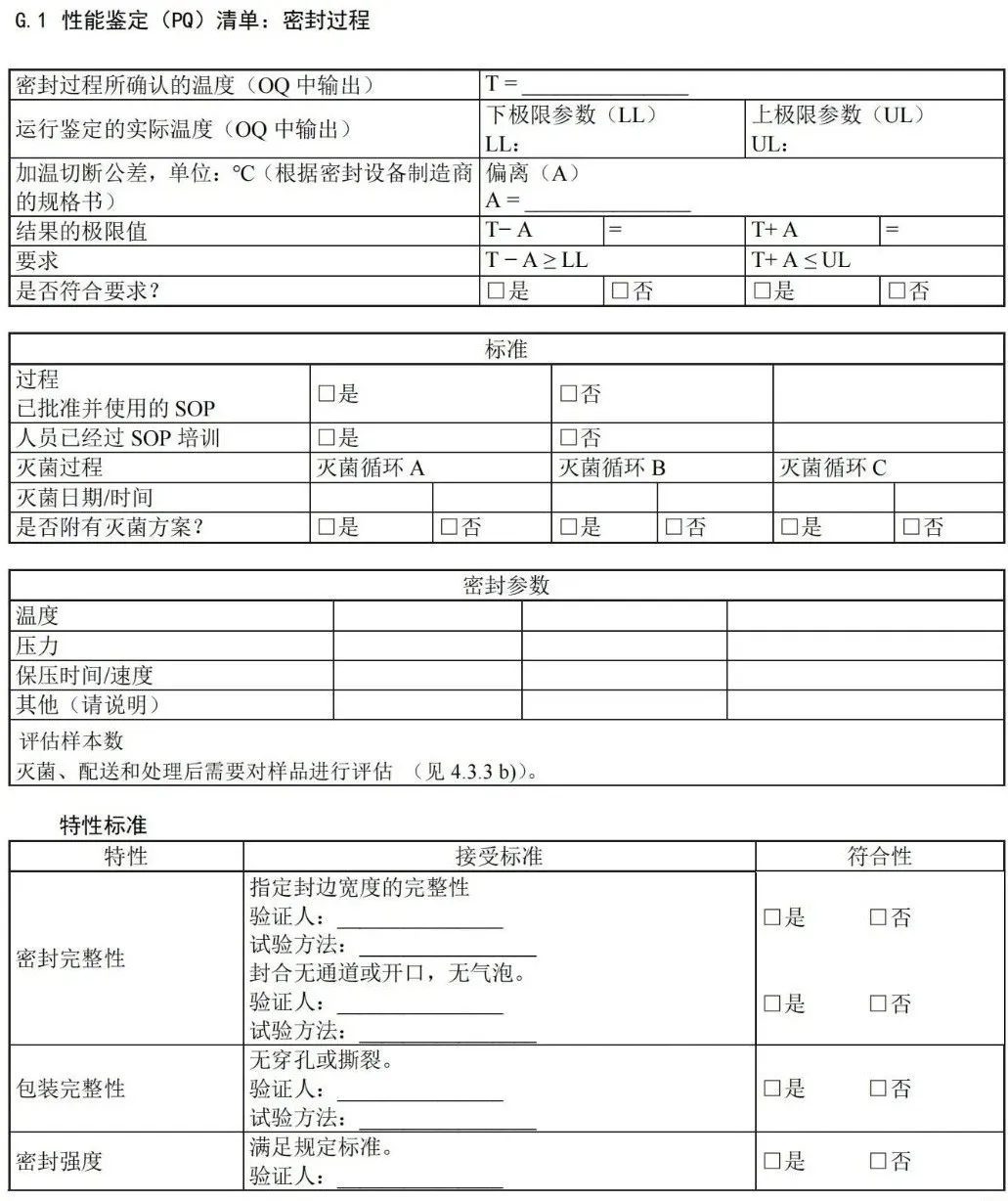
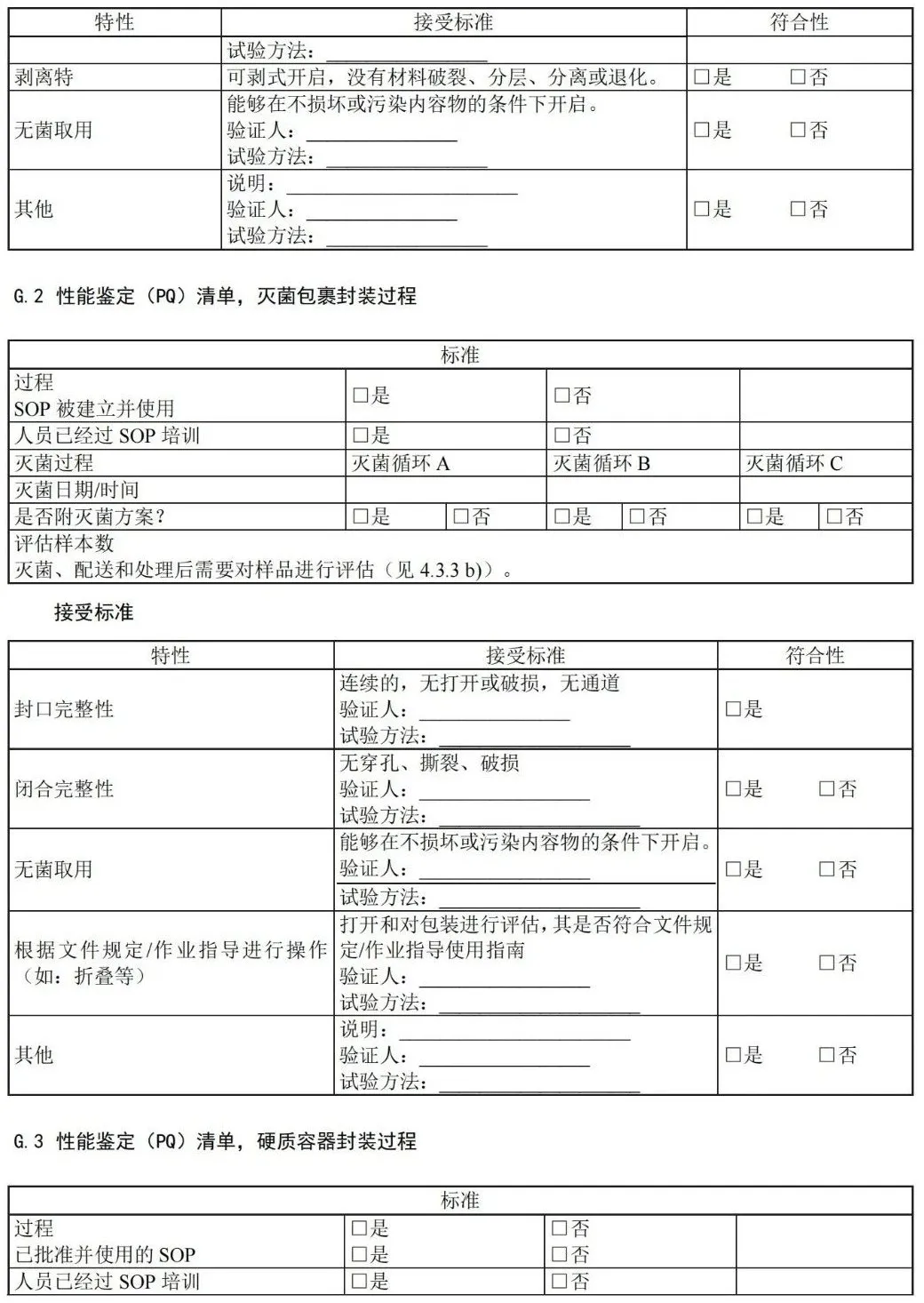
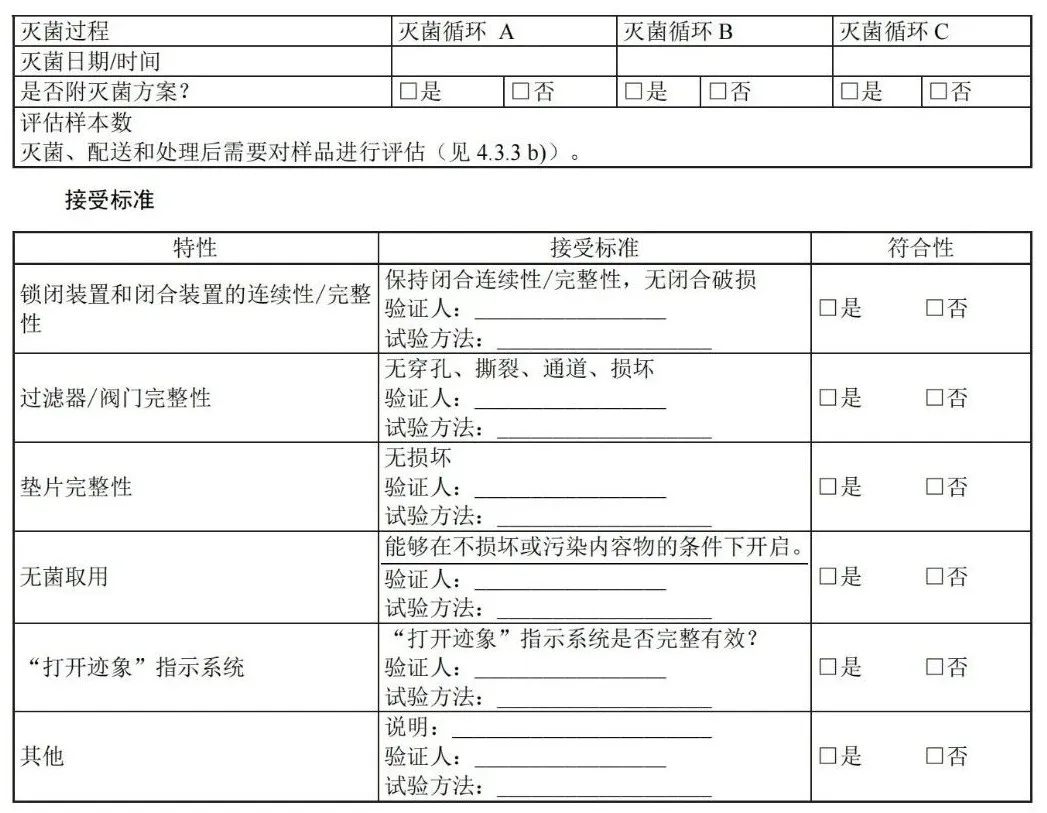
附 录 G (资料性) 性能鉴定文件——医疗机构指南
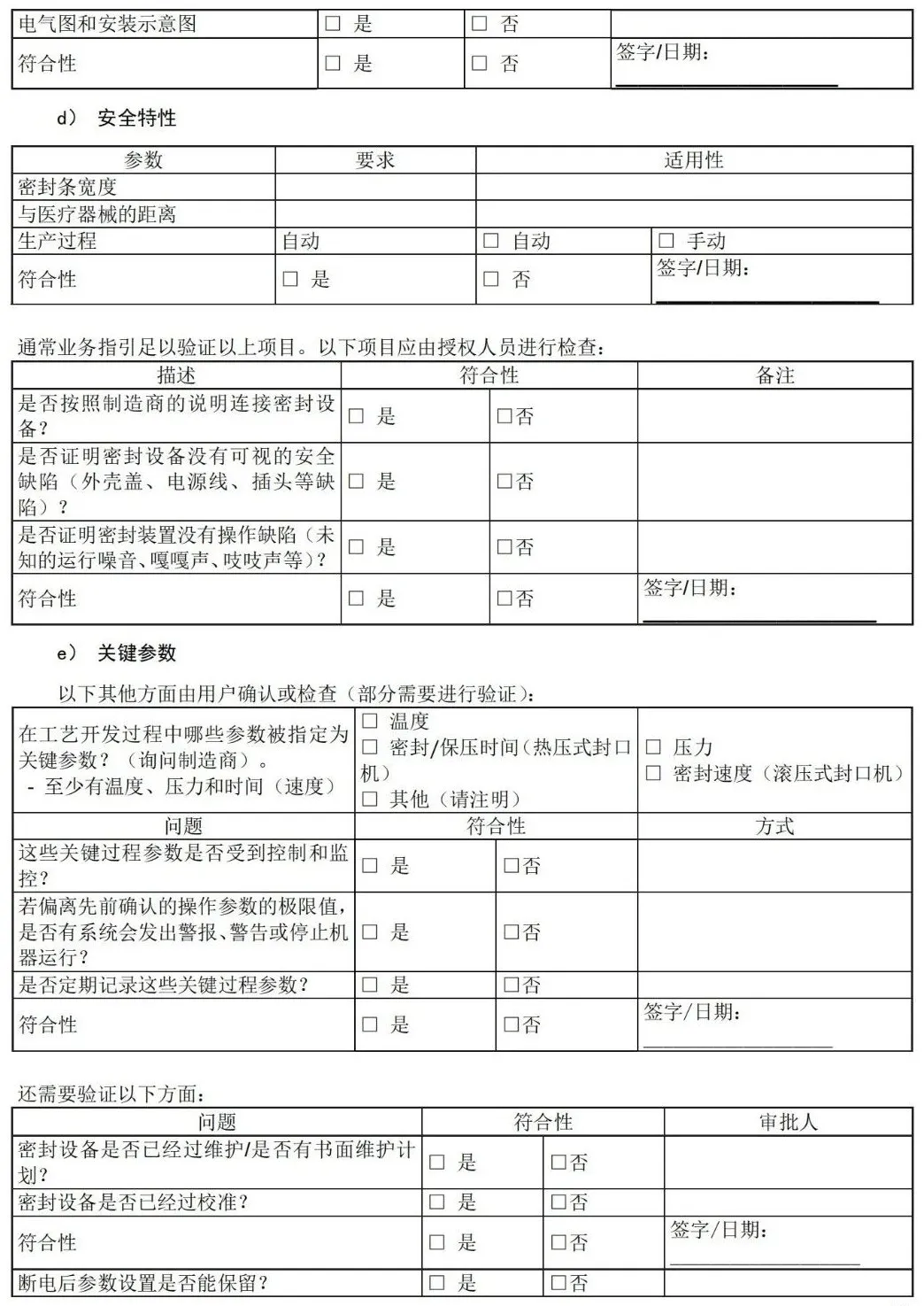
附 录 H (资料性) 应对最坏情况的要求——工业和医疗机构指南 H.1 概述 具体是指: ——无菌屏障系统(SBS):防止微生物进入并能使产品在使用地点无菌取用的最小包装; ——保护性包装:为防止无菌屏障系统和其内装物品从装配直到最终使用的时间段內受到损坏的材料结构; ——包装系统:无菌屏障系统和保护性包装的组合。 H.2 最坏情况构成 — 医疗器械 GB/T 19633.1-2015, 6.1.6 规定:“当相似的医疗器械使用相同的包装系统时,应对其结构相似性和最坏情况的识别加以说明并形成文件。至少应使用最坏情况的条件来确定是否符合本部分。” 在医疗器械产品族中(即类似但不完全相同的医疗器械),通常的无菌屏障系统可以用于保护各种医疗器械。通过对包装系统施加最大压力的医疗器械确定最坏情况。最坏情况构成可能是其他常见医疗器械组中最大或最重的产品,或者配件或其他医疗器械特征数目最多的产品。根据这些可清楚地确定最坏情况构成。但在某些情况下,可能需要测试多种类型的医疗器械(例如,最重的医疗器械和配件最多的医疗器械)以确保包装系统的最坏情况完全被挑战到。正确说明并评估最坏情况结构可以确保同产品系列的其他种类医疗器械能得到包装系统的适当保护。 H.3 最坏情况 — 无菌屏障系统 关于最坏情况试验的附加说明见 GB/T 19633.1-2015,6.3.4 包装系统性能试验相关规定,“性能试验应是在规定的成形和密封过程临界参数下,经过所有规定的灭菌过程后处于最坏状况下的无菌屏障系统上进行。”有两种主要方法说明本节所述的关键问题。 第一种最常见的方法是利用经充分确认的过程生产的预成形无菌屏障系统。在已确认的窗口内,常规操作条件下按批次生产的无菌屏障系统被测试并评估。通过从多个批次选择合适的样本量(通常为三个批次),可以确保在给定的置信水平下,代表了全部包装的特征(例如,密封强度)。因此,样本量和批次数量选择原理是确认文件的重要部分。许多现存参考资料有助于样本量的确定。 第二种方法是在最坏情况下生产无菌屏障系统,通常是采用经确认的工艺窗口的极限参数。在某些情况下,可在经确认的最低温度、最低压力和最短封合时间下生产出最差密封质量的无菌屏障系统。通常用 OQ 的参数生产的无菌屏障系统来评估包装系统的性能。这种方法代价高昂,需要在最坏情况下单独生产无菌屏障系统。 这两种方法,可使最终密封在达到或超出规定的过程极限参数下(不论是预成形无菌屏障系统、托盒/盖材系统,或者通过成型填充密封“FFS”过程),生产出最坏情况的无菌屏障系统。 医疗器械制造商符合 GB/T 19633.1 要求的方法不同,各种情况都应有合适的依据,并记录在包装确认方案中。方法的选择取决于公司风险管理方针及经济考量。 H.4 最坏情况构成 — 无菌屏障系统生产过程 GB/T 19633.2-2015,5.1.5 再次阐述了最坏情况构成,即“当确认相似的预成形无菌屏障系统和无菌屏障系统的制造过程时,确立相似性和最坏情况构型的说明应形成文件,至少应使最坏情况构型按本部分得到确认。”在这种情况下,最坏情况构成适用于无菌屏障系统生产过程,而不是医疗器械本身。当对生产过程进行确认时,可按产品族将(预成型)无菌屏障系统分组,例如包装材质相同但尺寸不同的V 型三边封组合袋。为确保整个无菌屏障系统产品族确认有效,需要识别出(预成型)无菌屏障系统系列的最坏情况构成。当采用加热密封时,须注意密封区的极端情况。例如,挑战并评估组合袋和吸塑包装上较小和较大封合区域的性能。对于热成型托盘和盖材,应考虑密封区域的排布总面积(即封合板下方的总密封面积)。这种方法可能产生过封和密封不足,需评估温度和压力分布的影响。 宜考虑到密封时的最坏情况包装构成可能与灭菌确认时的最坏情况不同。 附 录 I (资料性) 建立最终包装系统确认方案—工业指南
附 录 J (资料性) 设计输入 — 医疗器械特性 — 工业指南
附 录 K (资料性) 风险分析工具 —工业和医疗机构指南
附 录 L (资料性) 抽样计划注意事项 —医疗机构指南
附 录 M (资料性) 稳定性试验(GB/T 19633.1-2015, 6.4) — 工业指南 附 录 N (资料性) 互联网使用 — 工业和医疗机构指南 附 录 O (资料性) 试验方法确认 — 工业指南 附 录 P (资料性) 合同包装商的使用 — 工业和医疗机构指南
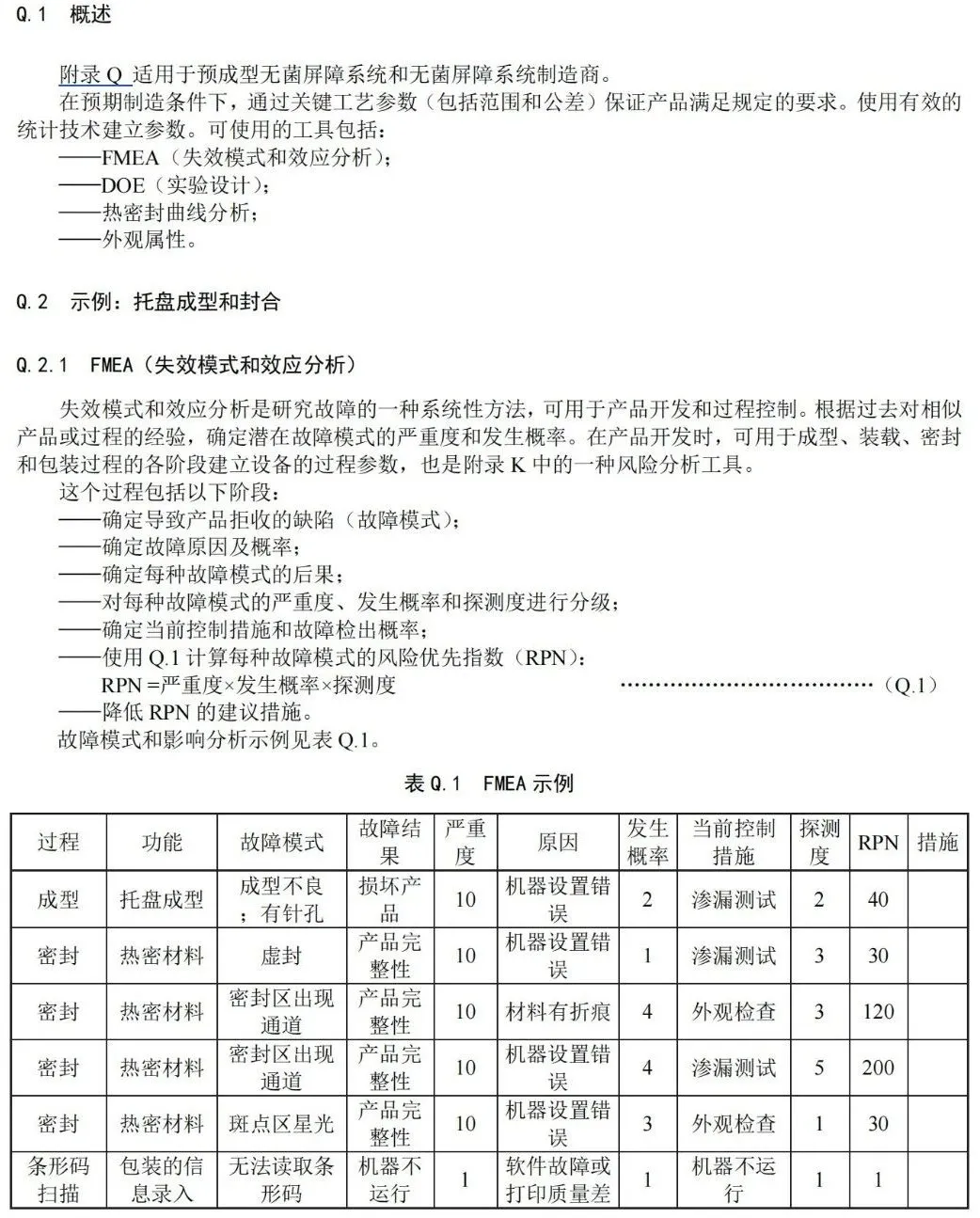
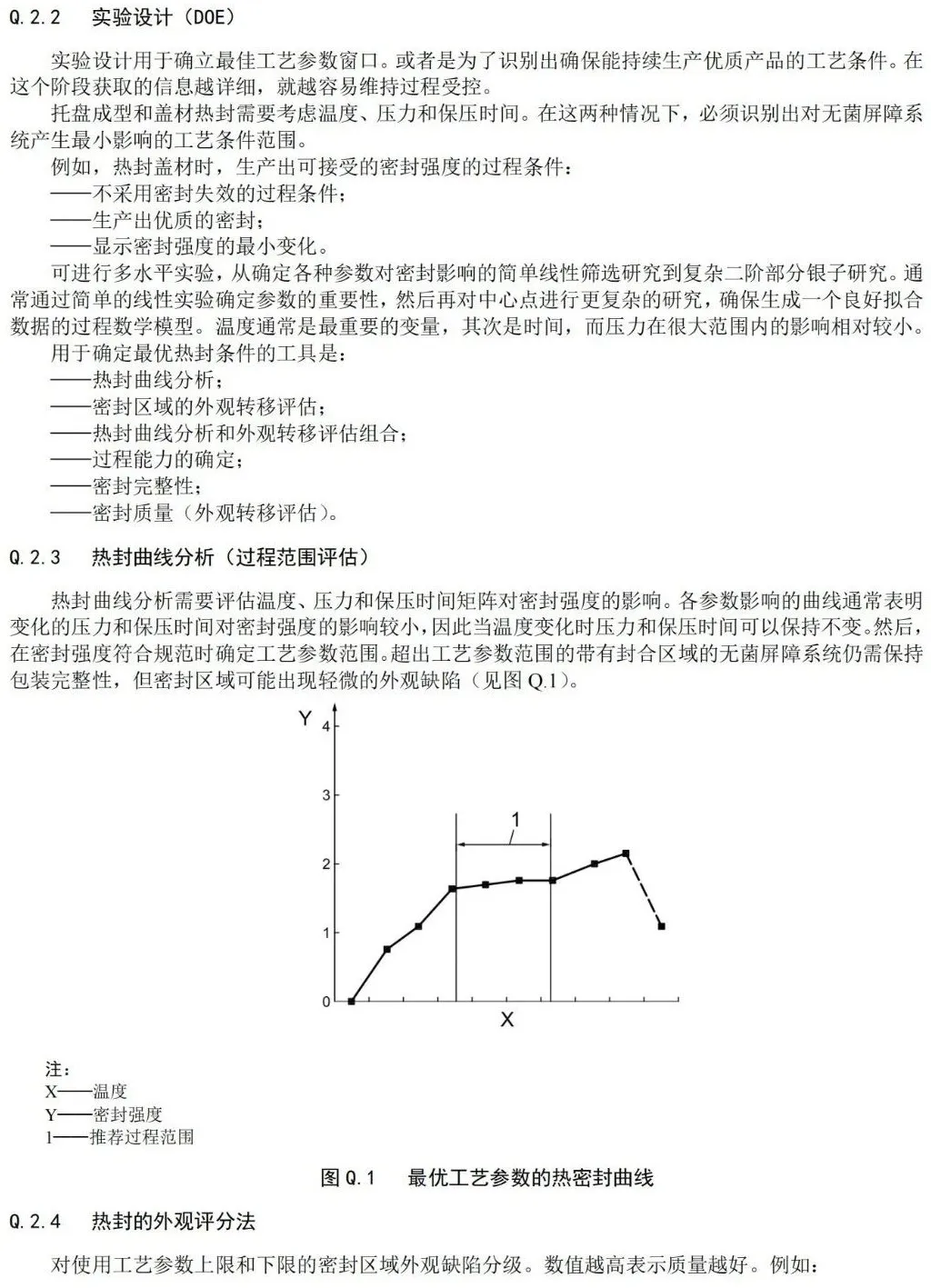
附 录 Q (资料性) 过程参数建立指南 — 工业指南
附 录 R (资料性) 故障调查 — 工业和医疗机构指南
附 录 S (资料性) 包装生产过程和包装系统设计可行性评估 — 工业指南
参考文献 [1]GB/T 450 纸和纸板 试样的采取及试样纵横向、正反面的测定 [2]GB/T 451.3 纸和纸板厚度的测定 [3]GB 2410 透明塑料透光率和雾度试验方法 [4]GB/T 2828.1 计数抽样检验程序 第 1 部分:按接收质量限(AQL)检索的逐批检验抽样计划 [5]GB/T 4669 纺织物 机织物 单位长度质量和单位面积质量的测定 [6]GB 8808 软质复合塑料材料剥离试验方法 [7]GB/T 8809 塑料薄膜抗摆锤冲击试验方法 [8]GB/T 9639.1 塑料薄膜和薄片 抗冲击性能试验方法 自由落镖法 第 1 部分:梯级法 [9]GB 9706.1 医用电气设备 第 1 部分:基本安全和基本性能的通用要求 [10]GB 10006 塑料薄膜和薄片摩擦系数测定方法 [11]GB/T 12914 纸和纸板 抗张强度的测定 恒速拉伸法(20 mm/min) [12]GB 13022 塑料 薄膜拉伸性能试验方法 [13]GB/T 15171 软包装件密封性能试验方法 [14]GB 16174.1 手术植入物 有源植入式医疗器械 第 1 部分:安全、标记和制造商所提供信息的通用要求 [15]GB 16174.2 手术植入物 有源植入式医疗器械 第 2 部分:心脏起博器 [16]GB/T 16578.1 耐撕裂性能的测定 第 1 部分:裤形撕裂法 [17]GB/T 16578.2 塑料 薄膜和薄片 耐撕裂性能的测定 第 2 部分:埃莱门多夫(Elmendor)法 [18]GB/T 16841 能量为 300 keV~25MeV 电子束辐射加工装置剂量学导则 [19]GB/T 16886.1 医疗器械生物学评价 第 1 部分:风险管理过程中的评价与试验 [20]GB/T 16886.7 医疗器械生物学评价 第 7 部分:环氧乙烷灭菌残留量 [21]GB 18278.1 医疗保健产品灭菌 湿热 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制 [22]GB 18279.1 医疗保健产品灭菌 环氧乙烷 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制的要求 [23]GB/T 18279.2 医疗保健产品灭菌 环氧乙烷 第 2 部分:GB 18279.1 应用指南 [24]GB 18280.1 医疗保健产品灭菌 辐射 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制要求 [25]GB 18280.2 医疗保健产品灭菌 辐射 第 2 部分:建立灭菌剂量 [26]GB/T 18280.3 医疗保健产品灭菌 辐射 第 3 部分:剂量测量指南 [27]GB/T 19633.1-2015 最终灭菌医疗器械包装 第 1 部分:材料、无菌屏障系统和包装系统的要求 [28]GB/T 19633.2-2015 最终灭菌医疗器械包装 第 2 部分:成形、密封和装配过程的确认的要求 [29]GB/T 19789 包装材料 塑料薄膜和薄片氧气透过性试验 库仑计检测法 [30]GB/T 19971-2015 医疗保健产品灭菌 术语 [31]GB/T 20220 塑料薄膜和薄片 样品平均厚度,卷平均厚度及单位质量面积的测定 称量法(称量厚度) [32]GB/T 25102.13 电声学 助听器 第 13 部分:电磁兼容(EMC) [33]GB/T 31995 医疗保健产品灭菌 辐射 证实选定的灭菌剂量 VDmax 方法 [34]YY/T 0316 医疗器械 风险管理对医疗器械的应用 [35]YY/T 0681.1 无菌医疗器械包装试验方法 第 1 部分:加速老化试验指南 [36]YY/T 0681.2 无菌医疗器械包装试验方法 第 2 部分:软性屏障材料的密封强度 [37]YY/T 0681.3 无菌医疗器械包装试验方法 第 3 部分:无约束包装抗内压破坏 [38]YY/T 0681.4 无菌医疗器械包装试验方法 第 4 部分:染色液穿透法测定透气包装的密封泄漏 [39]YY/T 0681.5 无菌医疗器械包装试验方法 第 5 部分:内压法检测粗大泄漏(气泡法) [40]YY/T 0681.6 无菌医疗器械包装试验方法 第 6 部分:软包装材料上印墨和涂层抗化学性评价 [41]YY/T 0681.7 无菌医疗器械包装试验方法 第 7 部分:用胶带评价软包装材料上印墨或涂层附着性 [42]YY/T 0681.8 无菌医疗器械包装试验方法 第 8 部分:涂胶层重量的测定 [43]YY/T 0681.9 无菌医疗器械包装试验方法 第 9 部分:约束板内部气压法软包装密封胀破试验 [44]YY/T 0681.11 无菌医疗器械包装试验方法 第 11 部分:目力检测医用包装密封完整性 [45]YY/T 0681.12 无菌医疗器械包装试验方法 第 12 部分:软性屏障膜抗揉搓性 [46]YY/T 0681.13 无菌医疗器械包装试验方法 第 13 部分:软性屏障膜和复合膜抗慢速戳穿性 [47]YY/T 0681.14 无菌医疗器械包装试验方法 第 14 部分:透气包装材料湿性和干性微生物屏障试验 [48]YY/T 0681.15 无菌医疗器械包装试验方法 第 15 部分:运输容器和系统的试验 [49]YY/T 0681.16 无菌医疗器械包装试验方法 第 16 部分:包装系统气候应变能力试验 [50]YY/T 0698(所有部分)最终灭菌医疗器械包装材料 [51]YY/T 0698.2 最终灭菌医疗器械包装材料 第 2 部分:灭菌包裹材料要求和试验方法 [52]YY/T 0698.5 最终灭菌医疗器械包装材料 第 5 部分:透气材料与塑料膜组成的可密封组合袋和卷材 要求和试验方法 [53]YY/T 0698.8 最终灭菌医疗器械包装材料 第 8 部分:蒸汽灭菌器用重复性使用灭菌容器 要求和试验方法 [54]YY/T 0884 适用于辐射灭菌的医疗保健产品的材料评价 [55]YY/T 1302.1 环氧乙烷灭菌的物理和微生物性能要求 第 1 部分:物理要求 [56]YY/T 1302.2 环氧乙烷灭菌的物理和微生物性能要求 第 2 部分:微生物要求 [57]YY/T 1402 医疗器械蒸汽灭菌过程挑战装置适用性的测试方法 [58]YY/T 1263 适用于干热灭菌的医疗器械的材料评价 [59]YY/T 1264 适用于臭氧灭菌的医疗器械的材料评价 [60]YY/T 1265 适用于湿热灭菌的医疗器械的材料评价 [61]YY/T 1266 适用于过氧化氢灭菌的医疗器械的材料评价 [62]YY/T 1267 适用于环氧乙烷灭菌的医疗器械的材料评价 [63]YY/T 1268 环氧乙烷灭菌的产品追加和过程等效 [64]YY/T 1276 医疗器械干热灭菌过程的开发、确认和常规控制要求 [65]YY/T 1608 医疗器械辐射灭菌 验证剂量实验和灭菌剂量审核的抽样方法 [66]YY/T 1613 医疗器械辐照灭菌过程特征及控制要求 [67]YY/T 1759 医疗器械软性初包装设计和评价指南 [68]T/CAMDI 033 医疗器械包装材料的生物学评价指南 [69]ISO 15747 Plastic containers for intravenous injections [70]ISO/TS 16775:2014 Packaging for terminally sterilized medical devices — Guidance on the application of ISO 11607-1 and ISO 11607-2 [71]ISO/TS 17665-2 Sterilization of health care products — Moist heat — Part 2: Guidance on the application of ISO 17665-1 [72]AAMI TIR19 Guidance for ISO 10993-7 Biological evaluation of medical devices—Part 7:Ethylene oxide sterilization residuals [73]AAMI TIR20 Parametric release for ethylene oxide sterilization [74]ANSI/ASQ Z1.4 Sampling Procedures and Tables for Inspection by Attributes [75]ANSI/ASQ Z1.9 Sampling Procedures and Tables for Inspection by Variables for Percent Nonconforming [76]ANSI/AAMI ST58 Chemical sterilization and high level disinfection in health care facilities [77]ANSI/AAMI ST63 Sterilization of healthcare products: Requirements for the development,validation and routine control of an industrial sterilization process for medical devices — Dry heat [78]ASTM D589 Standard test method for opacity of paper (15°diffuse illuminant A, 89% reflectance backing and paper backing) [79]ASTM D2457 Standard test method for spectacular gloss of plastic films and solid plastics [80]ASTM D4754 Standard test method for two-sided liquid extraction of plastic materials [81]ASTM D5264 Standard practice for abrasion resistance of printed materials by the Sutherland rub tester [82]ASTM F17 Standard terminology relating flexible barrier packaging [83]ASTM F372 Standard test method for water vapour transmission rate of flexible barrier materials [84]ASTM F1249 Standard test method for water vapour transmission rate through plastic film and sheeting [85]ASTM F1327 Standard terminology relating to barrier materials for medical packaging [86]ASTM F2029 Standard practices for making heatseals for determination of heatsealability of flexible webs as measure by seal strength [87]ASTM F2203 Standard test method for linear measurement using precision steel rule [88]ASTM F2251 Standard test method for thickness measurement of flexible packaging material [89]DIN 58953-7 Sterilization — Sterile supply — Part 7: Use of sterilization paper, nonwoven wrapping material, textile materials, paper bags and sealable pouches and reels [90]DIN 58953-8 Sterilization — Sterile supply — Part 8: Logistics of sterile medical devices [91]EN 13427 Packaging — Requirements for the use of European Standards in the field of packaging and packaging waste [92]EN 13428 Packaging — Requirements specific to manufacturing and composition — Prevention by source reduction [93]EN 13429 Packaging — Reuse [94]EN 13430 Packaging — Requirements for packaging recoverable by material recycling [95]EN 13431 Packaging — Requirements for packaging recoverable in the form of energy recovery,including specification of minimum inferior calorific value [96]EN 13432 Packaging — Requirements for packaging recoverable through composting and biodegradation — Test scheme and evaluation criteria for the final acceptance of packaging [97]IEST-STD-CC1246D Product Cleanliness Levels and Contamination Control Program [98]TAPPI Dirt estimation chart. Norcross (Ga.): TAPPI, 2000, Product code: 0109DIRT Norcross, GA [99]Dunkelberg H., MD; Schmelz , U., MD,. Determination of the Efficacy of Sterile Barrier Systems Against Microbial Challenges During Transport and Storage. Infect. Control Hosp. Epidemiol. 2009 February,30 (2) p. 179 [100]Fotis N., & Bix L. Sample Size Selection Using a Margin of Error Approach. Medical Device and Diagnostic Industry. 2015 October, 28 (10) pp. 80–89 [101]Sterilization Packaging Manufacturers Council (SPMC), Sterile Packaging: The Facts of Shelf Life in Medical Device Developments, Medical Device Network, March 2015 [102]Berry and Kohn’s Operating Room Technique, 9th Addition, 2000 by Nancymarie Fortunato 声明: 内容来源于行业标准信息服务平台,如有误或涉及版权问题请留言或联系,立即修正或删除。 转载自粤知感控 —————————————————— 为人类感控事业而奋斗——明誉医疗 推荐文章 |
|





















暂无留言